
Glomerular Disease Primer: Selected Glomerular Diseases
- Membranous Nephropathy
- Podocyte Diseases
- Minimal Change Disease
- Focal Segmental Glomerulosclerosis (FSGS)
- Collapsing Glomerulopathy
Membranous Nephropathy
Clinical Manifestations
Patients with membranous nephropathy may present with asymptomatic proteinuria, found on routine urinalysis performed as part of a physical examination, or may present with edema. Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin.
Pathology
Early in the course of disease, light microscopy may be nearly normal or may show thickening of the glomerular capillary loops. Immunofluorescence microscopy shows deposition of IgG (antibody) along capillary loops. Electron microscopy shows immune deposits (antibody) along the outside of the glomerular basement membrane, facing the urinary space and located underneath the podocyte).
Causes
Membranous nephropathy may be idiopathic (of unknown cause) or associated with infections (hepatitis B) or stem cell transplant.
Standard Therapy
Various immunosuppressive medications have been shown to induce remission in patients with membranous nephropathy, including steroids, cyclophosphamide, and cyclosporine. Edema is managed by dietary salt restriction and diuretics, either administered orally or intravenously.
Disease Course
Glomerular filtration rate is typically normal at the outset of membranous nephropathy, but in some patients glomerular filtration rate will decline after months or years. Risk factors for progressive loss of kidney function include heavy proteinuria and male sex.
NIH Research Studies
Patients who have not had prior therapy for membranous nephropathy, and those who have had steroids or cyclosporine, are eligible for an NIH clinical trial testing sirolimus. The study is open label, no placebo, and lasts up to 12 months.
Podocyte Diseases
Primary podocyte diseases can be classified by morphology and by etiology (cause).
| Disease | Genetic Cause | Acquired cause | Medication-association |
|---|---|---|---|
| Minimal change disease | Congenital presentation nephrin | Minimal change disease | NSAID (arthritis medicines) Gold injection Interferon-alpha Lithium Pamidronate |
| Focal segmental glomerulosclerosis | Congenital presentation Integrin beta4 Infancy/childhood presentation podocinPAX2WT1mitochondrial DNA Adult presentation Actinin-4 CD2AP TRPC6 mitochondrial DNA | Idiopathic FSGSPost-adaptive FSGS1) reduced kidney mass: congenital small kidneys, surgery, reflux nephropathy2) normal kidney mass: obesity, sickle cell disease | Cyclosporine Inteferon-alpha Lithium Pamidronate |
| Collapsing glomerulopathy | none proven | Idiopathic collapsing glomerulopathyHIV-associated nephropathy | Interferon-alpha Pamidronate |
Minimal Change Disease
Clinical Manifestations
Patients with minimal change typically present with the sudden onset of edema, either affecting the legs or, particularly in children, affecting the whole body, including facial edema (which may be the first manifestation). Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin. In children, the peak age of onset is age 2, with the disease being uncommon before age 1 and less common in during the teenage years. Minimal change disease also occurs in adulthood, even in the elderly, but is less common with advancing age.
Pathology
On light microscopy, the kidney appears normal or nearly normal. Older pathologic terms for this disease include nil disease (i.e, no abnormality) and lipoid nephrosis. Immunofluorescence microscopy may be normal or may show deposition of IgM antibody within the glomerulus. Electron microscopy shows podocyte foot process effacement, which is common to nearly all proteinuric diseases. Minimal change disease typically shows, prior to therapy, very extensive foot process effacement, often covering >90% of the glomerular capillary surface.
Causes
The cause or causes of minimal change disease are largely unknown. Occasionally, minimal change disease is associated with medication use (see table above) or may follow insect stings. Several lines of evidence suggest that the immune system contributes to the appearance of minimal change disease. This evidence includes 1) an increased prevalence of allergic disease tendency (atopy) in children with minimal change disease; 2) an increased prevalence of minimal change disease in people with lymphoma, a disorder of lymphocytes (an immune cell); 3) an increase in particular HLA genotypes, which indicate a tendency to particular patterns of immune responsiveness; 4) occasional remission of minimal change disease after particular viral infections, which activate the immune system. Nevertheless, the particular pathways by which the immune system might cause or promote minimal change disease are not understood.
Standard Therapy
Most children (>90%) and adults (>70%) enter a complete remission with a course of steroids. Mortality at present is very low. Many steroid regimens have been used. The most common regimens involve the use prednisone daily or every other day. Unfortunately, relapses are common. When patients are steroid-resistant (do not enter complete remission with steroid therapy), other therapeutic options include cyclosporine. When patients respond to steroids but experience frequent relapses, therapeutic options include cyclophosphamide, chlorambucil, and cyclosporine.
Edema is managed by dietary salt restriction and diuretics, either administered orally or intravenously.
Disease Course
Before the use of steroids became standard in the 1960’s, there was a high mortality in children with minimal change disease due to infection, as antibodies are lost in urine and this produces immunodeficiency. As discussed below, most patients respond to steroids and have an excellent long-term prognosis. In those who are steroid-resistant, a renal biopsy may subsequently show focal segmental glomerulosclerosis, which has a worse prognosis (more likely to progress to end-stage kidney disease). Spontaneous remissions (occurring without specific therapy) also occur.
NIH Research Studies
- As mentioned above, oral daily steroids are not effective for some patients with minimal change disease. The NIH Clinical Research Center in Bethesda, MD is sponsoring a trial of intermittent oral dexamethasone in adults who have received <8 weeks of prednisone and children who have received <10 weeks of prednisone. This trial is open-label, with every patient receiving active treatment. We are examining whether intermittent therapy will be as effective as standard daily therapy with less toxicity.
- Therapy-resistant minimal change disease, adults (proteinuria >3.5 g/d despite ACE inhibitor or ARB therapy): therapy with oral retinoic acids (alitretinoin or isotretinoin). Open label-study, no placebo, duration 24 week
Focal Segmental Glomerulosclerosis (FSGS)
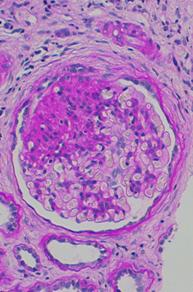 View full-sized image
View full-sized imageClinical Manifestations
Patients with focal segmental glomerulosclerosis (FSGS) may present in different ways. First some present with edema, either a sudden onset similar to that seen in patients with minimal change disease or slower onset over weeks to months. Second, some patients have no symptoms and are found to have asymptomatic (definition: no symptoms) proteinuria, found on routine urinalysis performed as part of a physical examination. Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin.
Pathology
The term focal segmental glomerulosclerosis is a pathologic term, referring to glomerulosclerosis (glomerular scarring, representing an increase in collagens and other proteins) that is found in a focal distribution (initially some glomeruli are affected while most glomeruli are entirely normal) and a segmental distribution (early in disease, the affected glomerular show sclerosis in a segmental pattern while other portions of that glomerulus remains normal). As the disease progresses, this pattern may shift so that some glomeruli are globally (totally) sclerosed and that all glomeruli have at least some sclerosis.
Causes
In the table above, we distinguish two broad forms of acquired FSGS. Idiopathic FSGS is of unknown cause and probably is the most common form. Post-adaptive FSGS arises after a phase in which the glomeruli are exposed to higher than normal blood flow for a number of years. Increased glomerular blood flow is typical of two settings. First, it occurs when the number of glomeruli is reduced, as happens following bilateral kidney surgery or congenital kidney abnormalities or chronic scarring of the kidney tubules (for example, due to reflux of urine from the bladder back into the kidney). Second, it occurs with obesity, sickle cell anemia and a few other conditions; the reasons for the increase in glomerular blood flow in these settings are unknown.
Occasionally other glomerular diseases, such as IgA nephropathy or membranous nephropathy or lupus nephritis may show focal and segmental glomerular scarring which may be referred by some pathologists as focal segmental glomerulosclerosis. These patients should be treated according to their underlying glomerular disease and the term FSGS is best not applied in these situations.
Most FSGS is sporadic (no family history). Some FSGS is familial (there is more than one affected family member). Genetic mutations may be seen in sporadic FSGS and familial FSGS, but is more common in the latter. To date eight genes have been associated with FSGS. This is an area of intense research and more genes are likely to be identified soon. Several inheritance patterns have been identified and the five most commonly-mutated genes will be discussed below.
- Autosomal recessive inheritance. The inheritance pattern skips generations; parents each carry one copy of the mutant gene and are themselves normal. In children with FSGS patients who also have a family history of FSGS, perhaps 35% will have two mutations in podocin. When families are small, podocin mutations can also appear as sporadic FSGS (no family history); perhaps 20% of children with sporadic FSGS will have podocin mutations. Reasons for pursuing a diagnosis of podocin mutation include the following: 1) steroid-resistance is typical; 2) the risk of FSGS recurrence after kidney translantation is lower than with idiopathic FSGS; 3) family counseling. Podocin mutations almost never cause adult-onset FSGS
- Autosomal dominant inheritance. This inheritance pattern affects every generation, with one parent and approximately 50% of children affected. Two genes are have been reported (alpha actinin-4 and TRPC6); in both cases FSGS appears during adulthood.
- Other patterns. WT-1 mutations cause FSGS appearing in the teenage years. Due to the effects of this mutation on sex development, these individuals may appear to be female but may lack ovaries and a uterus and consequently do not begin menstruation. Mitochondrial DNA mutations may be associated with isolated FSGS or the FSGS may be accompanied by other symptoms, include acidosis, stroke-like episodes, muscle weakness, deafness and diabetes.
Individuals of African descent are at increased risk for FSGS, by a factor of approximately 4-fold compared to those of other races and ethnic groups. It is likely that this has a genetic component but the specific gene or genes have not been identified.
Finally certain medications have been associated with FSGS.
Standard Therapy
Steroids are the therapy with the best chance of inducing a sustained complete remission in patients with FSGS; they may also induce a partial remission. While some patients with a complete remission experience relapse, they often undergo a second remission with a second course of therapy. Steroids may also induced a sustained partial remission. Standard courses of steroid therapy include daily or alternate day steroids for period of 2 to 4 months, possibly extended longer with a taper off if the response has been good.
For those who are steroid-responsive but experience one or more relapses when steroids are stopped, other therapies include cyclophosphamide or cyclosporine.
For those who are steroid-resistant, other therapies include cyclosporine or (less well studied) mycophenolate mofetil.
Edema is managed by dietary salt restriction and diuretics, either administered orally or intravenously.
Disease Course
At ten years after diagnosis, approximately 50% of individuals with FSGS will developed end-stage kidney disease, requiring dialysis or kidney transplant. This statistic includes both individuals who received treatment and did not respond favorably and individuals who came ot medical attention too late for effective therapy to be attempted. This prognosis makes FSGS the most serious primary glomerular disease. The prognosis in those who enter a complete remission (CR) is excellent, even when they experience a relapse. The long-term prognosis in those who enter a partial remission (PR) is less good, but is substantially better than those who did not respond to therapy or did not take therapy.
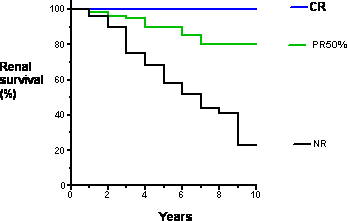 View full-sized image
View full-sized imageRecurrence After Kidney Transplant
Approximately 25% of patients who have FSGS in their own kidneys will experience recurrent FSGS after kidney transplant. When FSGS recurs, it typically does so within days to weeks of transplant, and essentially always within one year of transplant. The cause of recurrent FSGS is unknown but is believed to be a circulating protein. Patients at increased risk for recurrent FSGS include the following: rapid progression (from diagnosis to end-stage kidney disease in less than 3 years, recurrent FSGS in a prior transplant (risk of recurrent FSGS is approximately 70%), and white race (weak risk factor). In patients with FSGS, it is prudent to check the urine weekly by dipstick for protein or every 2 to 4 weeks by measuring a random urine protein/creatinine ratio. The appearance of proteinuria should prompt a transplant kidney biopsy, which would initially show just podocyte foot process effacement and after weeks to months of proteinuria will show FSGS. If the diagnosis can be made within 2 to 4 weeks of proteinuria onset, therapy may include plasma exchange and possibly cyclophosphamide.
NIH Research Studies
- Early diagnosis FSGS, children (proteinuria > 50 mg/kg) and adults (proteinuria >3.5 g/d): therapy with intermittent oral dexamethasone. Open-label study, no placebo, duration 48 weeks.
- Therapy-resistant FSGS, adults (proteinuria <3.5 g/d despite ACE inhibitor or ARB therapy): therapy with oral retinoic acids (alitretinoin or isotretinoin). Open label-study, no placebo, duration 24 week.
- FSGS with progressive decline in GFR despite ACE inhibitor or ARB therapy: therapy with oral pirfenidone. Open-label study, no placebo, duration 48 weeks, option to continue therapy longer if GFR decline rate is improved with therapy.
- Recurrent FSGS following kidney transplant: For those at high risk for recurrent FSGS, therapy involves plasma exchange prior to kidney transplant. For those who have experienced recurrent FSGS after kidney transplant and who have had proteinuria less than 4 weeks, therapy involves plasma exchange plus oral cyclophosphamide. Both trials are open-label, no placebo.
Collapsing Glomerulopathy
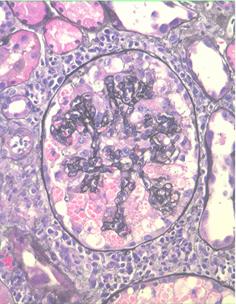 View full-sized image
View full-sized imageClinical Manifestations
Patients with collapsing glomerulopathy (also termed collapsing FGSGS) may present in different ways. First some present with edema, either a sudden onset similar to that seen in patients with minimal change disease or slower onset over weeks to months. Some patients describe an accompanying syndrome with some or all of the following symptoms: fever, chills, cough, diarrhea, and skin rash. Second, some patients have no symptoms and are found to have asymptomatic (definition: no symptoms) proteinuria, found on routine urinalysis performed as part of a physical examination. Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin.
Disease Course
Collapsing glomerulopathy has a course similar to FSGS but the average patient has a greater chance of progressing to end-stage kidney disease.
Pathology
Initially some or all glomeruli affected by collapse of the glomerular capillary tuft and proliferation of podocytes, which may accumulate within the urinary space. Later glomerular scarring develops, so that the pathology may come to resemble FSGS.
Causes
The major known cause is HIV-1 infection. Most patients with HIV infection and collapsing glomerulopathy are of African descent, suggesting that genetic factors contribute to susceptibility. Several other infections have been linked to collapsing glomerulopathy, including two tropical infections (loa loa and filiariasis). One infection whose association with collapsing glomeruopathy remains controversial is parvovirus B19 (the cause of a common pediatric infection, fifth disease or erythema infectiosum).
Standard Therapy
In general, therapy for collapsing glomerulopathy is similar to therapy for FSGS. Some nephrologists advocate an aggressive regimen that combines steroids and cyclosporine.
NIH Research Studies
- We are seeking patients with a new diagnosis of collapsing glomerulopathy for studies of viral causes; these studies are carried out using blood and urine samples.
- Early diagnosis collapsing glomerulopathy, children (proteinuria > 50 mg/kg) and adults (proteinuria >3.5 g/d): therapy with intermittent oral dexamethasone. Open-label study, no placebo, duration 48 weeks.
- Therapy-resistant collapsing glomerulopathy, adults (proteinuria <3.5 g/d despite ACE inhibitor or ARB therapy): therapy with oral retinoic acids (alitretinoin or isotretinoin). Open label-study, no placebo, duration 24 week.
- Collapsing glomerulopathy with progressive decline in GFR despite ACE inhibitor or ARB therapy: therapy with oral pirfenidone. Open-label study, no placebo, duration 48 weeks, option to continue therapy longer if GFR decline rate is improved with therapy.