

Small Molecule Stabilizers as a Potential Treatment for Transthyretin Familial Amyloid Polyneuropathy
Proteins are important building blocks for all body parts, including muscles, bones, hair, and nails, and perform vital biologic functions. Proteins circulate throughout the body in the blood and, when functioning properly, are beneficial. Occasionally, cells produce abnormal proteins that can misfold, form fibrillar deposits or aggregates and cause disease; the exact cause of fibril formation is unknown. When these deposits of abnormal proteins were first discovered, they were called amyloid, and the disease process was called amyloidosis. A common feature is that the deposits share a “β-pleated sheet” structural conformation with characteristic properties detectable by certain color dyes. Diagnosis of the various forms of amyloid disease is confirmed by tissue biopsy. In systemic amyloidosis, proteins produced in one part of the body travel to a different location where they become insoluble and form fibrillar deposits that impair organ function. One such form of systemic amyloid disease is transthyretin (TTR) amyloidosis. Past investments in basic science research have provided the foundation for an exciting small molecule approach to inhibit TTR amyloidosis.
The Blood Transport Protein Transthyretin in Health and Disease
TTR is a blood transport protein for the hormone thyroxine and the vitamin A-retinol binding protein complex. TTR has two binding sites for thyroxine but only a small percentage, perhaps less than 1 percent, of TTR in blood has thyroxine bound to it. The liver is the main site of TTR synthesis in the body, but TTR is also made in the retina and pancreas. Both mutant and normal forms of TTR can give rise to amyloid deposits. TTR consists of four identical subunits; scientists refer to the assembled subunits as a tetramer. Dissociation of the tetramer is the rate-limiting step for amyloid fibril formation. Any one of nearly 100 different mutations in the gene encoding TTR can cause amyloidosis. Disease-associated mutations destabilize the tetrameric structure and some increase the rate of tetramer dissociation. Specific clinical syndromes associated with mutated TTR are familial amyloid polyneuropathy (FAP) and familial amyloid cardiomyopathy. Senile systemic amyloidosis is a disorder that occurs in very elderly individuals (mostly men) in which amyloid fibrils formed from the normal TTR protein are deposited primarily in the heart, but also in the gut and in the carpal tunnel space of the wrist.
What is Transthyretin Familial Amyloid Polyneuropathy?
TTR FAP is a rare, progressive, and ultimately fatal hereditary neurodegenerative disease that affects the nerves and often the heart and kidneys as well. Symptoms include sensory loss, erectile dysfunction, alternating diarrhea and constipation, urinary incontinence, urinary retention, and delayed gastric emptying. TTR FAP is inherited in an autosomal dominant manner. The phrase “autosomal dominant” means that if one parent has the disease, there is a 50 percent chance that the disease gene will pass to a child. A mutation in TTR called “V30M” is the most common cause of FAP. Currently, there are no FDA-approved drugs to treat this rare but serious disease. Liver transplantation is a treatment option, as replacing a liver producing mutant TTR with a liver synthesizing normal TTR can slow, if not halt, disease progression. However, this approach has its limitations, which include matched donor availability, surgery, and a need for long-term immunosuppressive steroid treatment after the transplant.
Development and Characterization of TTR Small Molecule Stabilizers
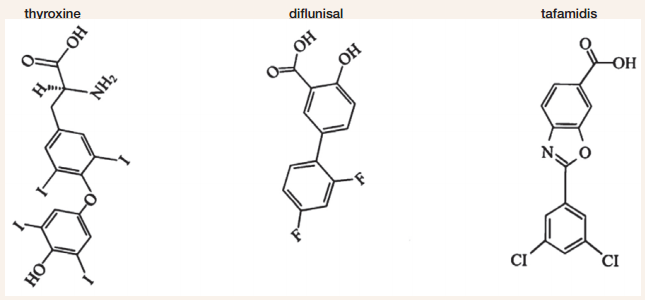
In the laboratory, the stability of TTR’s tetrameric structure can be assessed by placing it in a chemical known to disrupt or “denature” protein structures. In 1996, a strategy was developed to design an orally bioavailable small molecule stabilizer that binds to TTR in blood with both high affinity and selectivity, and prevents or significantly slows dissociation of the TTR tetramer. In a proof of principal experiment, thyroxine concentrations slightly higher than necessary to occupy all TTR binding sites stabilized both normal and mutant-containing (e.g., V30M) tetramers from dissociation under conditions of denaturation. Unfortunately, thyroxine cannot be used as a stabilizer of tetramer structure—owing to its hormone activity. However, results of this study did provide evidence that a small molecule having a similar structure to thyroxine without hormonal activity but with specific and selective binding affinity might effectively stabilize normal and mutant TTR. A related research study subsequently showed that occupancy of only one of the two thyroxine binding sites is sufficient to stabilize most TTR tetramers from dissociation under denaturing conditions.
Based on structural similarity to thyroxine, a small molecule called diclofenac was tested for its ability to stabilize TTR under denaturing conditions, and researchers also evaluated a set of 12 similar molecules. These were nonsteroidal anti-inflammatory small molecules (NSAISMs), and in 2002, several, including diclofenac, were shown to stabilize the normal TTR tetramer effectively, but they were less effective at stabilizing mutant TTR tetramers (e.g., V30M). In 2004, diclofenac and several additional NSAISMs were evaluated for their ability to stabilize tetramer structure of the most common disease-associated TTR variants, including V30M. This study demonstrated that the NSAISM diflunisal provided effective stabilization for the majority of the mutant variants and was more effective in this regard than diclofenac. Used as an FDA-approved non-steroidal anti-inflammatory drug for more than 2 decades, diflunisal has been commercially available in over 40 countries, including the United States.
Diflunisal was evaluated further for its ability to selectively bind and stabilize 1) FAP variant TTR in blood samples, and 2) TTR when orally administered to healthy volunteers or patients with FAP. When diflunisal was added to the blood of FAP patients at potentially therapeutic concentrations, mutant V30M TTR tetramers in the blood were significantly stabilized under denaturing conditions, more so than the TTR of healthy volunteers. Already FDA-approved for mild to moderate pain, fever, and inflammation, orally administered diflunisal at 250 mg twice daily for 7 days was shown to stabilize TTR in blood samples obtained from a small pilot study of patients with FAP. Moreover, a second pilot study of orally administered diflunisal at 250 mg or 500 mg twice daily for 7 days showed that the drug occupied at least 1 of the 2 thyroxine binding sites and stabilized TTR tetramer structures from healthy volunteers. Published in 2006, the results of these two pilot studies suggested that diflunisal might be an effective small molecule therapeutic for treating TTR amyloidosis. In 2003, a series of compounds called benzoxazoles, which also have a structure similar to thyroxine, were synthesized, and 11 of 28 were shown to effectively stabilize normal TTR tetramers and to prevent amyloid fibril formation under denaturing conditions. One of these analogs, tafamidis, exhibited particularly favorable binding selectivity and stabilization of the normal TTR tetramer. Because of these findings, tafamidis was selected for additional pre-clinical and clinical research studies. As reported in 2012, tafamidis was found to effectively stabilize tetramers of the most clinically significant mutant form of TTR (V30M) associated with FAP, such that it behaves like normal TTR under denaturing conditions. Occupancy of one of the thyroxine binding sites by tafamidis stabilized 67 percent of normal TTR tetramers from dissociation under denaturing conditions. Increasing tafamidis concentrations such that both thyroxine binding sites were occupied stabilized 97 percent of the TTR tetramers. Consistent with previous results, tafamidis was also shown to selectively bind and stabilize TTR in human blood. When added to blood from patients with FAP at concentrations sufficient to occupy one or both of the thryoxine binding sites, tafamidis was found to significantly stabilize the V30M TTR under denaturing conditions. Furthermore, tafamidis was shown to stabilize a broad range of other pathogenic TTR variants in blood; these TTR variants contained mutations called Y69H, F64S, I84S, L111M, or V122I.
Clinical Trial Assessments of Diflunisal and Tafamidis
These two small molecule stabilizers have now been studied in clinical trials to test their safety and efficacy. Boston University has sponsored a placebo-controlled multicenter phase III clinical trial testing the efficacy of diflunisal for the treatment of FAP, and results are expected to be reported in 2013. This trial, with a target enrollment of 140 participants, has compared 250 mg diflunisal taken orally twice daily with a placebo. Results of a separate clinical trial evaluating tafamidis were reported by the Scripps Research Institute in July 2009. This 18-month phase II/III clinical trial, conducted by a pharmaceutical company co-founded by Dr. Kelly, showed that tafamidis slowed the progression of FAP in patients with the V30M TTR mutation. Further testing is now ongoing to achieve U.S. Food and Drug Administration approval of tafamidis for the treatment of TTR FAP.
NIDDK-supported Translational Research
The translation of scientific knowledge and technology into improvements in the practice of medicine is central to the missions of the NIH and the NIDDK. As this story illustrates, the clinical understanding of the pathological underpinnings of TTR amyloidosis spurred NIDDK support of basic science research that led to the development of small molecule stabilizers that effectively decrease TTR tetrameric dissociation and amyloid fibril formation. Once identified and characterized, these small molecules were well positioned to attract academic and industry interest in conducting clinical trials of these potential therapeutics. The NIDDK continues to be committed to supporting innovative strategies for improving the health of patients with FAP and many other diseases.