Herman K. Edskes, Ph.D.

Professional Experience
- Research Fellow, NIH, 2000-2001
- Postdoctoral Fellow, NIH, 1995-1999
- Ph.D., University of Kentucky, 1994
- M.Sc., Wageningen University, 1990
Research Goal
Studies of non-Mendelian elements in yeast have allowed the first proof of the prion concept, the existence of infectious proteins. Building upon these discoveries, we are working to further detail prion formation. We are also working to elucidate cellular responses to the presence of prions. This work facilitates studies of analogous mammalian systems.
Current Research
Prions
Using the genetically tractable yeast Saccharomyces cerevisiae, we are studying prions. Prions are infectious proteins. They can transmit a disease or a trait without an accompanying nucleic acid. The idea that proteins can form infectious agents is derived from studies of the transmissible spongiform encephalopathies (TSEs) in mammals. TSEs are fatal neurodegenerative diseases. Examples are scrapie of sheep, mad cow disease, chronic wasting disease of deer and elk, and kuru and Creutzfeldt-Jakob disease in humans. At least 10 proteins in S. cerevisiae are capable of forming a prion. Most of these proteins acquire their infectious nature through the formation of a type of ordered linear protein aggregate called amyloid. However, not all yeast prions are based on protein aggregation. Amyloid deposits are also found in people suffering from Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and late-onset diabetes.
Ure2 protein and the [URE3] prion
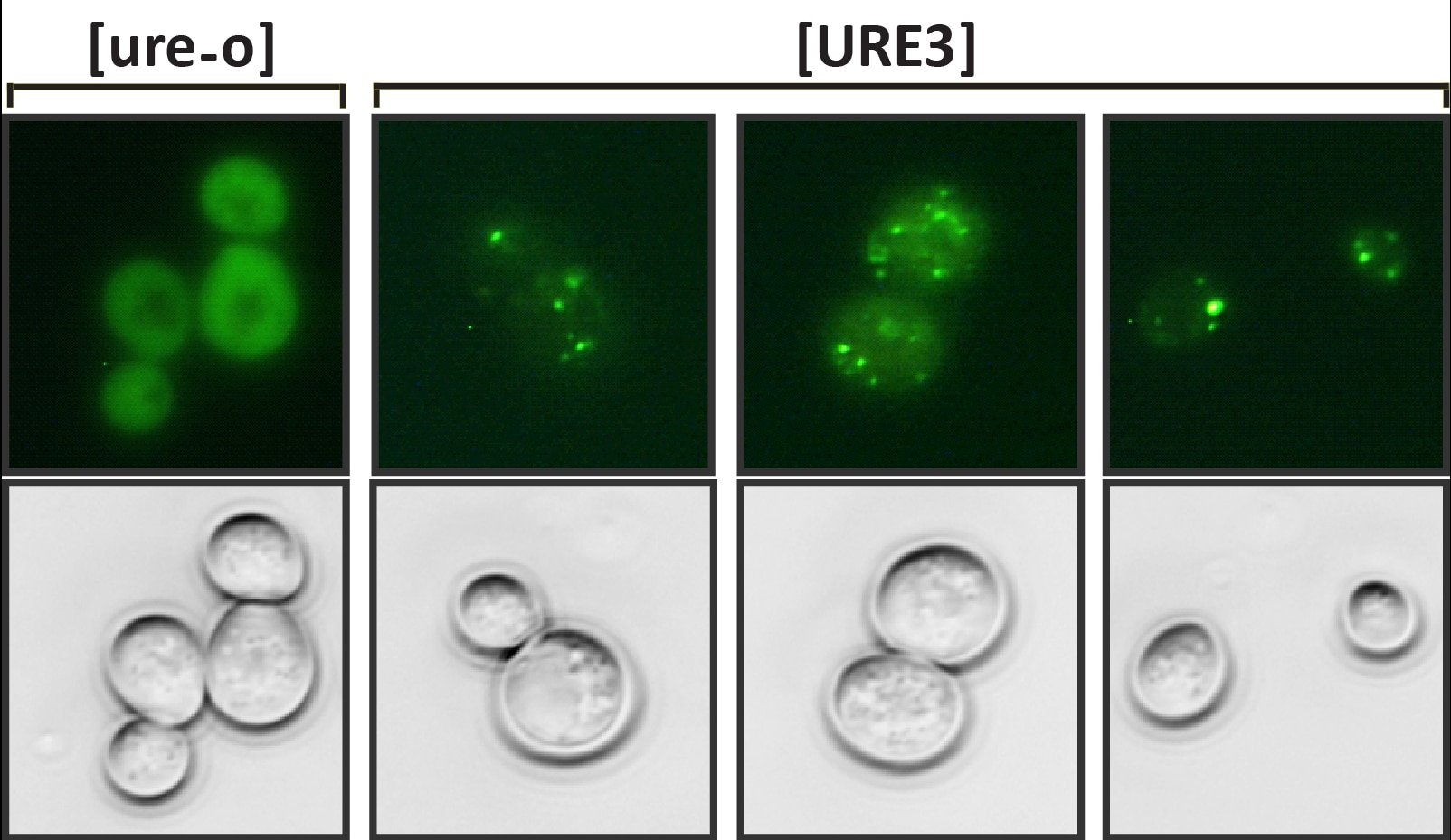
We have focused our attention mainly on the [URE3] prion (the brackets indicate its nonchromosomal inheritance). The prion arises spontaneously in a population at a frequency of approximately one to 10 per million cells. Overexpression of Ure2 protein dramatically increases the frequency with which [URE3] is found in a population. The normal function of Ure2 protein is to turn off the uptake and utilization systems of nitrogen sources that don’t support optimal growth when good nitrogen sources are available. This nitrogen-response system, often referred to as nitrogen catabolite repression, is found in many fungi and bacteria. The C-terminal domain of the protein resembles glutathione S-transferases and like glutathione S-transferases the Ure2 protein is a dimer. The asparagine-rich N-terminal domain of 94 amino acids is natively unstructured and is needed for the normal stability of the full-length protein. It forms amyloid in vitro and is sufficient for [URE3] propagation. As such, this domain is called the prion domain. Introduction of Ure2 amyloid into yeast results in cells with [URE3], thus this prion is an infectious amyloidosis of the Ure2 protein.
Amyloid and [URE3] variants
One of the remarkable features of [URE3] is that many stably propagating variants exist. These variants can be identified through differences in propagation efficiency, effect on engineered reporters and effect on yeast growth. [URE3] variants can also dramatically differ in infection ability. We learned this when we replaced URE2 with homologues from ascomycete yeasts in S. cerevisiae. A [URE3] variant from one Ure2 protein could only infect S. cerevisiae expressing the same protein. However, another variant from the same Ure2 protein could infect S. cerevisiae expressing Ure2 proteins from any of several yeasts. Thus, infection barriers exist that depend on the protein sequence and the prion variant. Similar infection barriers have been reported for TSEs. For instance, a variety of mammals, including humans, have been infected with mad cow disease, but there are no reports of people having been infected with sheep scrapie.
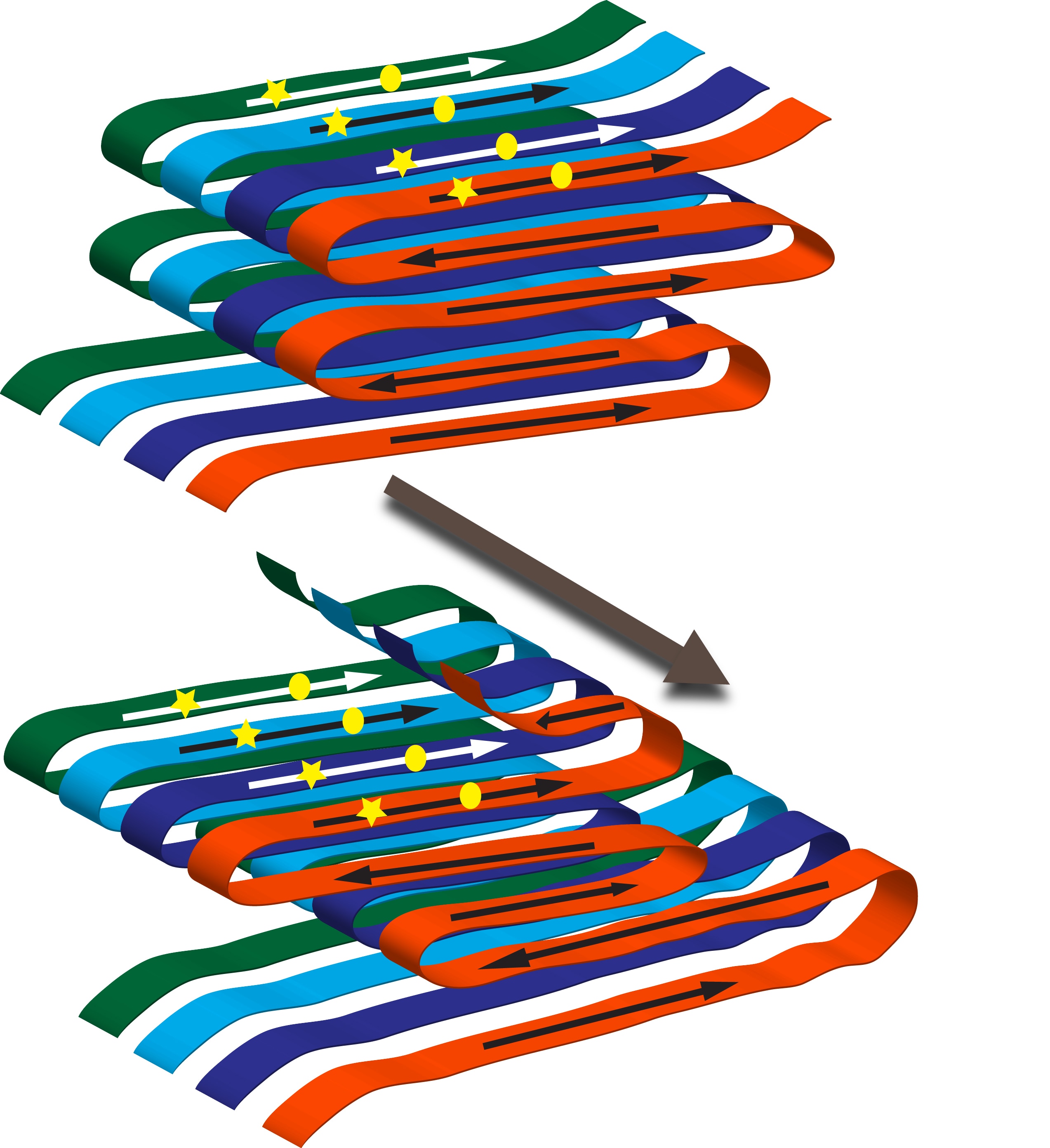
How can one protein form several prion variants? In amyloid filaments the N-terminal domain of Ure2 protein forms parallel in-register beta-sheets with the beta-strands perpendicular to the long axis of the filaments. Thus in the sheets the same amino acid is lined up in adjacent molecules. These sheets are folded multiple times along the long axis of the filament. Variants arise from different folds of the sheets as well as from the sheets being at different positions along the peptide chain. A monomer newly joining the end of the filament will assume the same fold as the molecules already in the fiber.
Ure2 proteins of yeasts
Ure2 protein homologues from ascomycete yeasts all have a C-terminal glutathione S-transferase like domain. When these protein sequences are aligned the N-terminal domains show much greater variation than the C-terminal domains. Even within one species, amino acid substitutions are more prevalent in the prion domain of Ure2 protein. This variation restricts the spread of [URE3] as it affects optimal alignment of a newly joining molecule with the existing Ure2 proteins in the amyloid filament. We showed that the Ure2 proteins of Naumovozyma castellii and the human pathogenic yeast Candida glabrata, despite the presence of a prion-like domain, are not capable of forming a prion in S. cerevisiae. The Candida glabrata protein also does not form amyloid in vitro. Both proteins fulfill the nitrogen regulatory function when they replace Ure2 protein in S. cerevisiae. Together these results indicate that the N-terminal domains of Ure2 proteins are not primarily evolved to form prions.
[URE3] a prion disease in yeast
Newly de novo formed [URE3] is sometimes poorly transferred to the daughter cell. However, once established, [URE3] is reliably propagated. If [URE3] were a benefit to cells carrying it, we would expect to find the prion in wild populations. A survey of 70 wild Saccharomyces isolates did not identify any that contained [URE3]. The same survey identified wild isolates that contained viruses and plasmids not known to provide any advantage to the cells. In addition, the [PIN+] prion was found with the same frequency as viruses and plasmids. Thus [URE3] does not provide the cells a net benefit.
Cells containing newly de novo formed [URE3] often form small colonies. Upon propagation, colony size becomes more comparable to that of uninfected cells. This suggests that initially, [URE3] negatively affected the cells but then changed to a variant with a more neutral effect on the cells. The absence of [URE3] in wild Saccharomyces isolates and the presence of unstable toxic variants points to this prion as a disease of yeast.
Current work
Although the prion concept has been firmly established in yeast, there are still many unresolved issues. The mechanisms of prion formation, replication, and transmission are only vaguely understood. Likewise, many questions remain about the interactions between prions and their host cells. We are further exploring prion formation of Ure2 and other proteins.
Applying Our Research
Humans are of great biological complexity. It has proven far easier to study human biology using model organisms than it is to discover de novo using human cells how the various systems work. The simple eukaryote Saccharomyces cerevisiae allows genetic manipulation at a level not available in any other organism. Fundamental biological processes show remarkable conservation between humans and S. cerevisiae. Studies of the transmissible spongiform encephalopathies provided a framework for the idea that proteins can form infectious elements. However, it was work with S. cerevisiae that proved the existence of infectious proteins (prions).
Strains of mammalian prions have puzzled scientists since their first report in 1961. Work with prion forming proteins from S. cerevisiae provided a solution to this puzzle. Some of these proteins fold into linear aggregates, called amyloid, by forming parallel in-register beta-sheet structures. Many variations of these structures can be formed by one protein thus explaining prion variants. Amyloid is not only found in the transmissible spongiform encephalopathies, it is associated with more than 20 human diseases including Alzheimer’s disease and Parkinson’s disease.
Studies of amyloid forming proteins in yeast will help our understanding of how cellular machineries affect amyloids and how amyloids affect cellular machineries. This will advance our understanding of these human diseases. In addition to the amyloid based prions, S. cerevisiae also contains prions formed through other mechanisms. There are still many diseases of unknown etiology and it is conceivable that studies of prions in yeast will shed light on some of these. Work with S. cerevisiae will continue to shed light on important biological problems.
Need for Further Study
We have only started to explore mechanisms by which proteins can become infectious elements. Many discoveries have yet to be made. The spectrum of proteins capable of forming a prion has not been mapped yet. General methods need to be developed to search for new prions. Of special importance is the identification of non-amyloid based prions. The mechanisms by which amyloid based prions are formed, replicated, and transmitted are still poorly understood. Likewise, the interaction between these prions and the cell are not completely clear and will require much more work. Knowledge gained from these studies will further our understanding of the dynamics of biological systems. Many diseases are still without known causes and are awaiting explanation and treatment.
Select Publications
- Innate immunity to prions: anti-prion systems turn a tsunami of prions into a slow drip.
- Wickner RB, Edskes HK, Son M, Wu S, Niznikiewicz M.
- Curr Genet (2021 Dec) 67:833-847. Abstract/Full Text
- Proteasome Control of [URE3] Prion Propagation by Degradation of Anti-Prion Proteins Cur1 and Btn2 in Saccharomyces cerevisiae.
- Edskes HK, Stroobant EE, DeWilde MP, Bezsonov EE, Wickner RB.
- Genetics (2021 May 17) 218. Abstract/Full Text
Research in Plain Language
Starting in the late 1980s and continuing into the late 1990s, many newspapers featured front-page stories about mad cow disease (or bovine spongiform encephalopathy). An unusual agent causes the infectious mad cow disease. This agent is a misfolded protein, not a virus or a bacterium. Such proteins are called prions (pronounced pree-ons).
Mad cow disease belongs to a group of prion diseases called transmissible spongiform encephalopathies (TSEs). TSEs are fatal diseases that break down cells of the brain and nervous system. TSEs also include the human diseases kuru and Creutzfeldt-Jakob disease. In TSEs, enzymes no longer break down a particular protein. As a result, these proteins bunch together through a process called protein aggregation. This process results in a substance called an amyloid. In humans, TSE diseases are rare, but amyloid deposits occur in much more prevalent diseases, such as Alzheimer’s and Parkinson’s. Health experts and the public have expressed concern about mad cow disease transmission to people.
Mammals are not the only organisms with prions. These infectious proteins also occur in fungi. In 1994, Dr. Reed Wickner showed that the [URE3] element of the yeast Sacchromyces cerevisiae (S. cerevisiae) is a prion of the Ure2 protein. The [URE3] prion is an infectious form of amyloid comprised from Ure2 protein. Scientists have demonstrated that several more proteins form a prion in S. cerevisiae. Most of these prions result from a yeast protein that forms amyloid. However, not all prions are based on amyloid formation.
Both mammalian and fungal prions can form strains with distinct biological properties. These are called variants for the fungal prions. In amyloid filaments, the Ure2 protein folds in a particular way to form a structure called a sheet. Variants arise from different folds of the sheets and from the sheets at different positions. To visualize this, imagine pouring syrup on a pancake. You make a pattern going from top to bottom, while at the same time making left and right movements. Cut away all parts of the pancake not covered with syrup. Make several exact copies and stack them on top of each other. This is your amyloid filament. When you vary the left and right movement, it is easy to see different patterns. These represent different amyloids.
Our research group studies [URE3] and other yeast prions. We try to understand how proteins form infectious elements. We are also very interested in the cellular machinery that facilitates prion formation. Another area of research examines how prions affect cells. Studying yeast prions enhances our understanding of similar infectious elements present in animals and people.
Research Images