

Definition & Facts of Biliary Atresia
In this section:
- What is biliary atresia?
- Are there different types of biliary atresia?
- How common is biliary atresia?
- Who is more likely to have biliary atresia?
- What are the complications of biliary atresia?
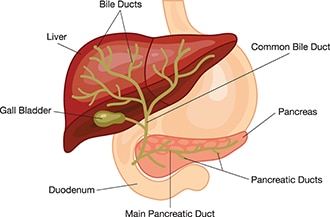
What is biliary atresia?
Biliary atresia is a condition in infants in which the bile ducts—tubes inside and outside the liver—are scarred and blocked. Bile ducts carry bile from the liver to the gallbladder for storage, and to the first part of the small intestine, also called the duodenum, for use in digestion. In infants with biliary atresia, bile can’t flow into the intestine, so bile builds up in the liver and damages it. The damage leads to scarring, loss of liver tissue and function, and cirrhosis.
Biliary atresia is life-threatening, but with treatment, most infants with biliary atresia survive to adulthood.
Are there different types of biliary atresia?
Doctors have identified different types of biliary atresia.
Biliary atresia without birth defects
In the most common type of biliary atresia, infants have no other major birth defects. Doctors may call this type of biliary atresia perinatal or isolated biliary atresia. A recent North American study found that 84 percent of infants with biliary atresia have this type.1
Biliary atresia with birth defects
Some infants have major birth defects—including problems with the heart, spleen, or intestines—along with biliary atresia. Doctors may call this fetal or embryonic biliary atresia. A recent North American study found that 16 percent of infants with biliary atresia have major birth defects.1
How common is biliary atresia?
Biliary atresia is rare and affects about 1 out of every 12,000 infants in the United States.2
Who is more likely to have biliary atresia?
Biliary atresia only occurs in newborn infants. The disease is slightly more common in female infants and in infants with Asian or African American heritage.3
What are the complications of biliary atresia?
Complications of biliary atresia include failure to thrive and malnutrition, cirrhosis and related complications, and liver failure.
Without treatment, infants with biliary atresia would develop cirrhosis within 6 months and liver failure within 1 year.3,4 By age 2, untreated infants would need a liver transplant to survive.4
Early treatment with a surgery called the Kasai procedure may slow or, in some cases, prevent the development of cirrhosis and liver failure. Even with treatment, about half of children with biliary atresia will need a liver transplant by age 2. Two-thirds will need a liver transplant sometime during childhood.3
Malnutrition
Even after treatment with the Kasai procedure, children with biliary atresia may have reduced bile flow to the small intestine and liver damage, leading to malnutrition and related problems with growth, such as failure to thrive. Read more about how biliary atresia affects nutrition.
Cirrhosis and related complications
Cirrhosis is a condition in which the liver breaks down and is unable to work normally. Scar tissue replaces healthy liver tissue, partly blocking the flow of blood through the liver. In the early stages of cirrhosis, the liver continues to work. As cirrhosis gets worse, the liver begins to fail.
In children with biliary atresia, cirrhosis may cause complications, including portal hypertension. Portal hypertension is high blood pressure in the portal vein, a blood vessel that carries blood from the intestines to the liver.
Portal hypertension may lead to specific complications, including
- a buildup of fluid in the abdomen, called ascites. Infection of this fluid can be very dangerous.
- enlarged blood vessels, called varices, which can develop in the esophagus, stomach, or both. Varices can break open and cause life-threatening bleeding in the digestive tract.
Liver failure
With liver failure, also called end-stage liver disease, the liver can no longer perform important functions or replace damaged cells. Infants and children with liver failure need a liver transplant to survive.
References
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases
(NIDDK), part of the National Institutes of Health. NIDDK translates and disseminates research findings to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by NIDDK is carefully reviewed by NIDDK scientists and other experts.
The NIDDK would like to thank:
Benjamin L. Shneider, M.D., Baylor College of Medicine, Texas Children’s Hospital