

Biliary Atresia
View or Print All Sections
Definition & Facts
Biliary atresia is a condition in infants in which the bile ducts outside and inside the liver are scarred and blocked. Bile can’t flow into the intestine, so bile builds up in the liver and damages it. The damage leads to scarring, loss of liver tissue and function, and cirrhosis.
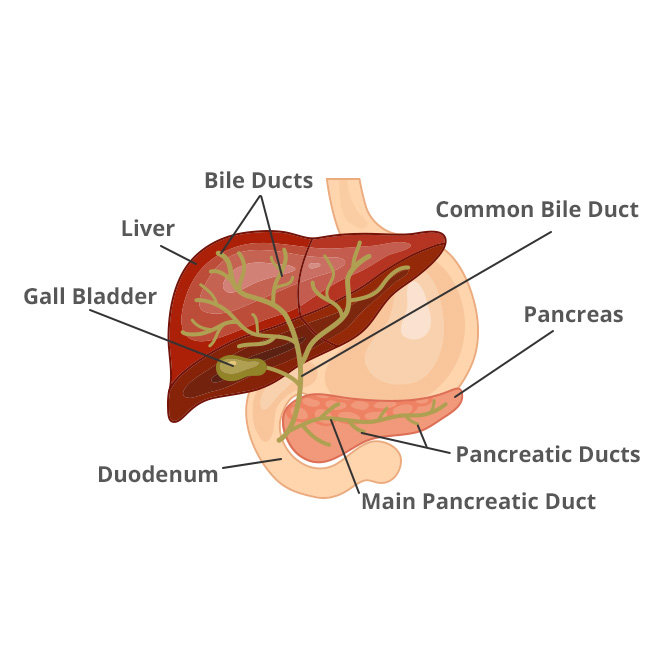
Symptoms & Causes
Typically, the first sign of biliary atresia is yellowing of the skin and whites of the eyes, called jaundice, which results from the buildup of bile in the body. Infants with biliary atresia typically develop jaundice by 3 to 6 weeks of age. Experts don’t know what causes biliary atresia.
Diagnosis
To diagnose biliary atresia, doctors will ask about the infant’s medical and family history, perform a physical exam, and order a series of tests. If test results suggest that an infant is likely to have biliary atresia, the next step is surgery to confirm the diagnosis.
Treatment
Doctors treat biliary atresia with a surgery called the Kasai procedure and eventually, in most cases, a liver transplant. Thanks to advances in treatment, more than 80 to 90 percent of infants with biliary atresia survive to adulthood.
Eating, Diet, & Nutrition
Children with biliary atresia may have reduced bile flow to the small intestine and liver damage, which may lead to malnutrition. To make sure infants and children with biliary atresia get enough nutrients and calories, doctors may recommend a special eating plan and supplements.
Clinical Trials
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and other components of the National Institutes of Health (NIH) conduct and support research into many diseases and conditions.
Related Conditions & Diseases
Related Diagnostic Tests
View More Liver Disease Information
Related Research
See more about liver diseases research at NIDDK.
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases
(NIDDK), part of the National Institutes of Health. NIDDK translates and disseminates research findings to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by NIDDK is carefully reviewed by NIDDK scientists and other experts.
The NIDDK would like to thank:
Benjamin L. Shneider, M.D., Baylor College of Medicine, Texas Children’s Hospital