

Acromegaly
On this page:
- What is acromegaly?
- How common is acromegaly?
- Who is more likely to develop acromegaly?
- What are the complications of acromegaly?
- What are the symptoms of acromegaly?
- What causes acromegaly?
- How do doctors diagnose acromegaly?
- How do doctors treat acromegaly?
- Clinical Trials for Acromegaly
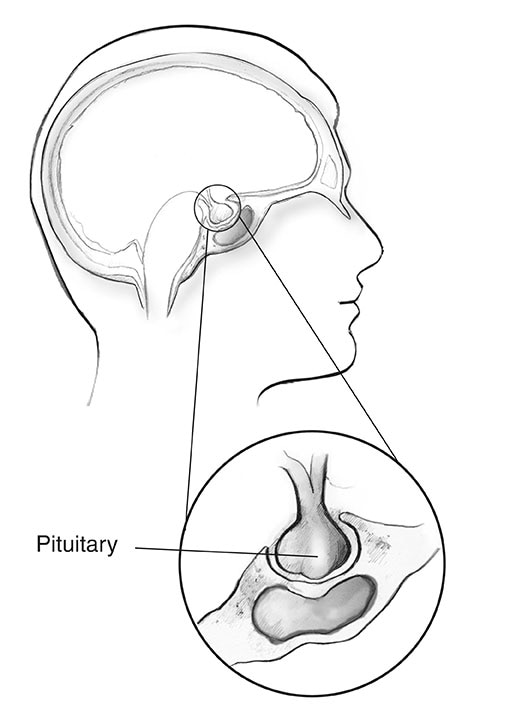
What is acromegaly?
Acromegaly is a disorder that occurs when your body makes too much growth hormone (GH). Produced mainly in the pituitary gland, GH controls the physical growth of the body. In adults, too much of this hormone causes bones, cartilage, body organs, and other tissues to increase in size. Common changes in appearance include enlarged or swollen nose, ears, hands, and feet.
How common is acromegaly?
Acromegaly is rare. Scientists estimate that about 3 to 14 of every 100,000 people have been diagnosed as having acromegaly.1
Who is more likely to develop acromegaly?
Acromegaly is most often diagnosed in middle-aged adults, but symptoms can appear at any age. In children, too much growth hormone causes a condition called gigantism rather than acromegaly. Gigantism occurs when excess GH begins before the end of puberty, when children’s growth plates fuse or close. Having too much GH before the growth plates close causes children to grow tall in height.
What are the complications of acromegaly?
Acromegaly is treatable in most people. But because symptoms come on slowly, health problems can develop before the disorder is diagnosed and treated.
Health problems can include
- type 2 diabetes
- high blood pressure
- heart disease
- sleep apnea
- arthritis
- carpal tunnel syndrome
- other conditions affecting the bones and muscles
People with acromegaly also have an increased risk for colon polyps, which may develop into colon cancer if not removed.
Some people with acromegaly may have a genetic condition that can lead tumors to develop in different parts of their bodies. Increased GH can cause these other tumors to grow.
Untreated, acromegaly can lead to serious health problems and early death. But when successfully treated, symptoms generally improve and may go away altogether. Life expectancy may return to normal.2
What are the symptoms of acromegaly?
Symptoms of acromegaly can vary from person to person. Common changes in physical appearance include
- hands and feet become larger and swollen—you may notice a change in ring or shoe size, especially shoe width
- lips, nose, and tongue become larger
- bone changes: brow and lower jaw jut out, bridge of the nose gets bigger, and space between teeth increases
- skin becomes thick, coarse, and oily
- sweating and skin odor increase
- voice becomes deeper
- skin tags—small, usually flesh-colored growths of skin that have a raised surface—may get larger or darker
Other common symptoms include
- headaches
- joint aches
- vision problems
What causes acromegaly?
Acromegaly develops when the pituitary gland releases too much GH into the body over a long period of time. When GH enters the blood, this signals the liver to produce another hormone, called insulin-like growth factor I (IGF-I). IGF-I is the hormone that actually causes bones and body tissue to grow. High levels of this hormone also cause changes in how the body processes blood glucose (blood sugar) and lipids (fats), which can lead to type 2 diabetes, high blood pressure, and heart disease.
In more than 9 out of 10 cases, acromegaly is caused by a tumor in the pituitary gland, called a pituitary adenoma.3 More rarely, the cause may be a tumor in another part of the body.
Although scientists don’t know what causes these tumors to develop, genetic factors may play a role. In young adults, acromegaly has been linked to defects in certain genes.
Pituitary tumors
Pituitary tumors are almost always benign, or noncancerous. Some tumors grow slowly, and symptoms of too much GH may not be noticed for many years. Other tumors may grow rapidly.
Depending on its size and location, the tumor may press against other pituitary tissue. Possible effects include
- changes in menstruation in women
- erectile dysfunction in men
- changes in thyroid hormone, which can affect weight, energy levels, hair, and skin
- decreases in cortisol, which can cause weight loss, dizziness, tiredness, low blood pressure, and nausea
A tumor that grows large in size may also press against nearby parts of the brain. This can lead to other symptoms, such as headaches and vision problems.
Some pituitary tumors that create growth hormone can also increase the levels of other hormones in the body. For example, the tumor may produce prolactin, the hormone that prompts the mammary glands to produce milk. This can lead to breast milk discharge in women.
Nonpituitary tumors
Rarely, acromegaly is caused by tumors located in the hypothalamus—a small area of the brain near the pituitary gland, pancreas, lungs, or other parts of the chest or abdomen. Some of these tumors make growth hormone themselves. But more often, the tumors produce growth hormone-releasing hormone (GHRH), a hormone that signals the pituitary gland to make growth hormone.
How do doctors diagnose acromegaly?
Blood tests
Doctors most often diagnose acromegaly by ordering two blood tests that help determine if your body is making too much GH.
- IGF test. Levels of GH in the blood can change throughout the day. A reliable way to track GH in the body is by measuring the level of IGF-I in the blood. In most cases, a high IGF-I level suggests that you have acromegaly.
- Oral glucose tolerance test. To confirm the diagnosis, your doctor will order an oral glucose tolerance test. For this test, you will drink a sugary liquid. A health professional will then test your blood every half hour for 2 hours to measure growth hormone levels. The sugar in the drink will normally cause GH levels to fall. But if your body is making too much of the hormone, these levels will not go down enough—thereby confirming the diagnosis of acromegaly.
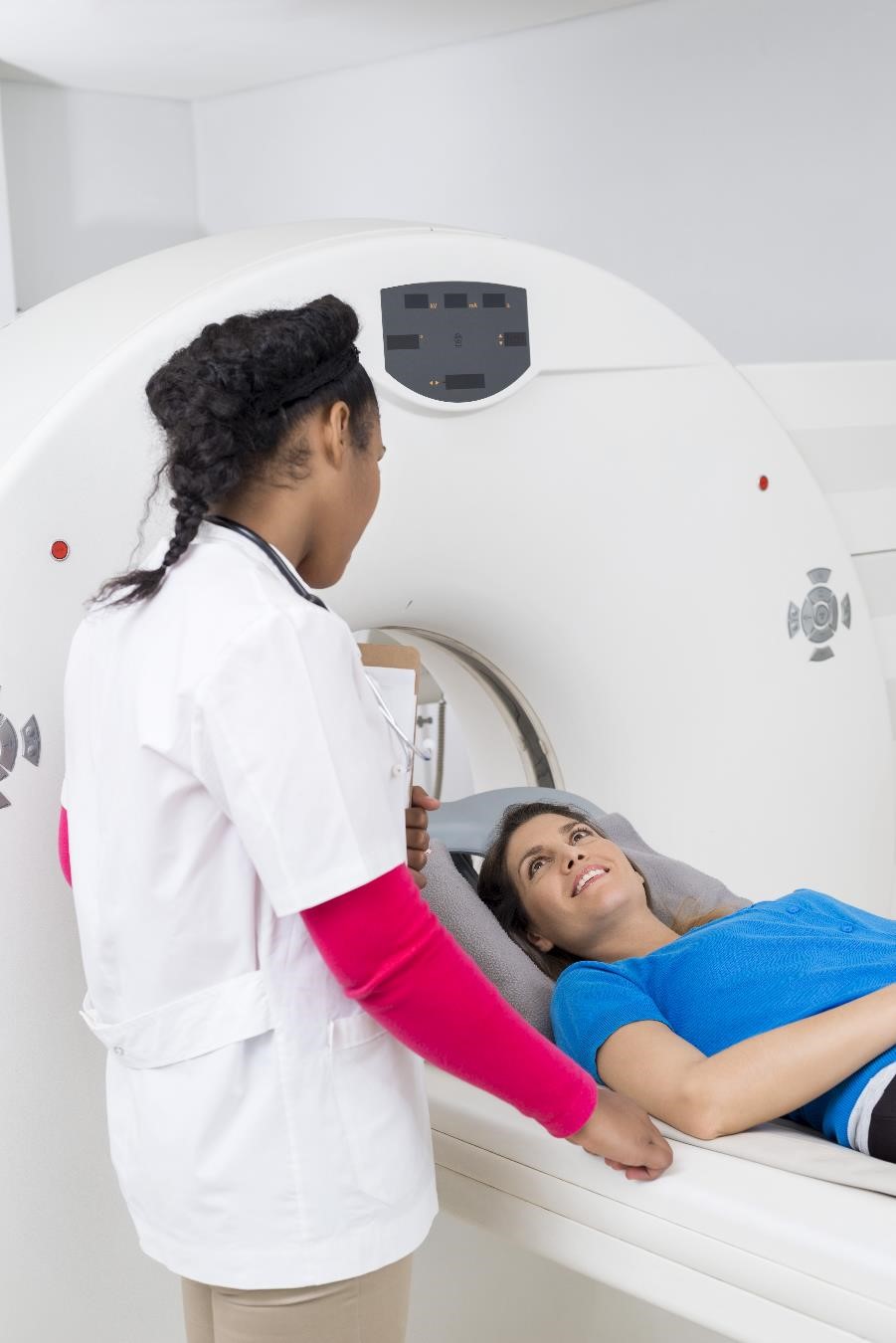
Imaging tests
If the blood tests confirm that your body is making too much GH, your doctor will conduct imaging tests to locate and measure the tumor that may be causing the problem. Two commonly used tests are
- Magnetic resonance imaging. The preferred test for viewing a pituitary tumor is the magnetic resonance imaging (MRI) scan. The MRI scan uses radio waves and magnets to create detailed images of your internal organs and soft tissues without x-rays.
- Computed tomography scan. If an MRI is not a good option for you (for example, if you have a pacemaker or other implant that has metal), your doctor may order a computed tomography (CT) scan instead. The CT scan uses a combination of x-rays and computer technology to create images of your organs and other internal parts of your body.
If the imaging test doesn’t find a pituitary tumor, your doctor will look for nonpituitary tumors as the cause of your high GH levels.
How do doctors treat acromegaly?
Treatment options include surgery, medicines, and radiation therapy. The goals of treatment are to control tumor size, return GH and IGF-I levels back to normal, improve symptoms, and manage related health problems. No single treatment is right for everyone. Your doctor will recommend a treatment plan that works for you, depending on factors such as your age, tumor size, severity of symptoms, GH and IGF-I levels, and health status.
Surgery
Doctors can remove most pituitary tumors using a method called transsphenoidal surgery. The operation is done through the nose and sphenoid sinus, a hollow space in the skull behind the nasal passages and below the brain. Two approaches to this surgery are
- with a microscope—a magnifying tool
- with an endoscope—a thin, lighted tube with a tiny camera
In both approaches, the surgeon uses advanced MRI imaging to scan the area around the tumor before surgery. He or she then makes a small cut inside your nostril to view the area and remove the tumor using tiny, special tools. In microscopic surgery, the surgeon uses a microscope to magnify the area. In endoscopic surgery, an endoscope camera sends images to a television monitor instead. Risks and results are similar for both approaches.3
When the tumor that is creating too much GH is not located in the pituitary gland, other types of surgery are used to remove the tumor. Removing these nonpituitary tumors also lowers GH levels and improves acromegaly symptoms.
Risks. Complications from surgery can include bleeding, cerebrospinal fluid leaks, meningitis, sodium (salt) and water imbalance, and low levels of pituitary hormones.3
Outcomes. The surgery is considered a success if blood levels of GH and IGF-I return to normal after 12 weeks. The cure rate right after surgery is about 85 percent for small tumors and 40 to 50 percent for large tumors.3
When successful, the surgery relieves pressure on nearby areas of the brain and causes GH levels to drop right away. Soft tissue swelling may get better within a few days but facial changes may take longer to improve.
Surgery is most successful in people with smaller pituitary tumors. Success largely depends on the skill and experience of the surgeon, as well as the location of the tumor. Even experienced surgeons may not be able to remove the tumor if it’s too close to parts of the brain where surgery would be risky. However, surgeons may be able to remove part of the tumor.
Postsurgery treatments. In most cases, levels of GH and IGF-I improve but don’t go back to normal. If levels of these hormones are still too high or begin to rise again, you may need further treatment. Most often, this will involve taking medicines. In some cases, your doctor may recommend a second surgery.
Medicines
Currently, three types of medicines are used to treat acromegaly, but they are not a cure. The medicines may be used alone or in combination with each other.
Somatostatin analogs. The medicines most often used to treat acromegaly are called somatostatin analogs (SSAs). These drugs curb the release of GH and may also reduce the size of the pituitary tumor. Several studies have shown that these drugs are safe and effective for long-term treatment. The medicines are delivered by injection, but scientists are currently studying other options, such as pills.4 The most common side effects of SSAs are cramps, gas, and diarrhea. These effects are usually mild and go away over time. Some people may develop gallstones that usually do not cause symptoms. Hair loss is possible and, in rare cases, permanent. Control of blood sugar usually improves but, rarely, may worsen.
Dopamine agonists. These medicines inhibit GH production and tumor growth, but not as well as SSAs do. Dopamine agonists are most likely to work in people who have mild GH excess and those who have both acromegaly and hyperprolactinemia (too much of the hormone prolactin). The medicines are taken by mouth. Side effects can include nausea, stuffed nose, tiredness, headache, dizziness when standing, nightmares, and mood changes.
Growth hormone-receptor antagonists. Unlike the other two medicines, GH-receptor antagonists do not stop the body from making too much GH. Instead, they block GH from signaling the body to make more IGF-I. The drug is taken in the form of a daily injection under the skin that patients can administer themselves. Side effects can include liver problems.
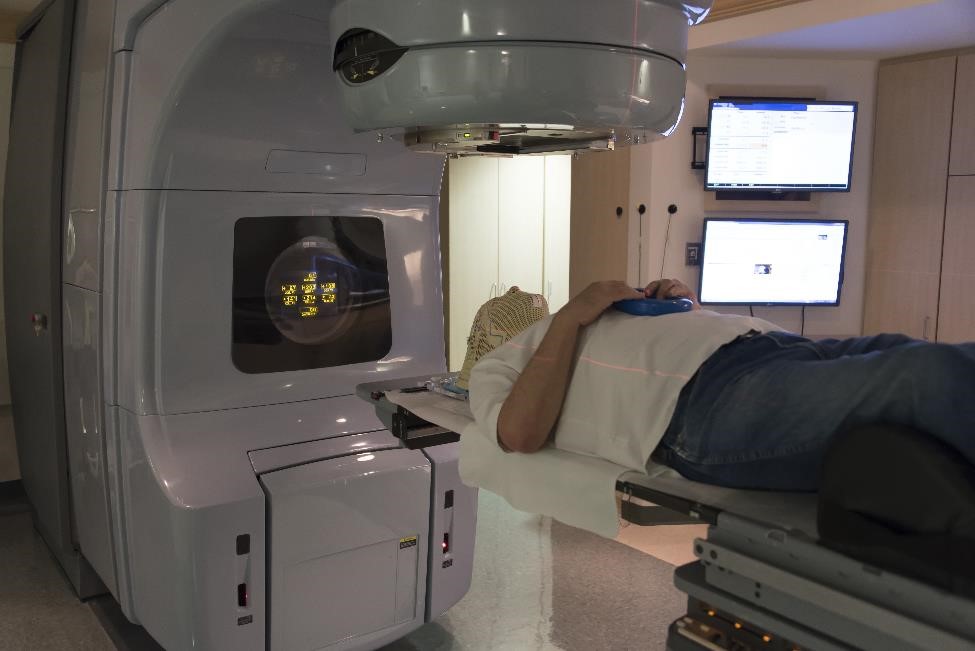
Radiation therapy
The third treatment option is radiation therapy, which uses high-energy x-rays or particle waves to kill tumor cells. This type of treatment may be recommended if surgery isn’t possible or fails to remove all tumor tissue, and medicines are not an option or working for you.
Stereotactic. The preferred type of radiation therapy is stereotactic radiation therapy, which uses 3-D imaging to precisely aim high doses of radiation to the tumor from various angles.3 The treatment can sometimes be done in a single session, reducing the risk of damage to nearby tissue. However, a single dose may not work for very large tumors and tumors located close to nerves that affect vision.
Conventional. The second option is conventional radiation therapy, which also targets the tumor with external beams. This type of radiation therapy delivers small doses of radiation in a series of treatments over 4 to 6 weeks.
As radiation treatment lowers GH and IGF-I levels over time, it may take years for this treatment to noticeably improve acromegaly symptoms. Your doctor is likely to prescribe medicines while you wait for GH and IGF-I levels to go back to normal and for symptoms to improve.
All forms of radiation therapy cause other pituitary hormones to slowly decrease over time. About half of people treated with radiation therapy will need hormone replacement after treatment ends. Radiation can also impair a patient’s fertility.
Vision loss and brain injury are rare complications. Rarely, other types of tumors can develop many years later in areas that were in the path of the radiation beam.
Clinical Trials for Acromegaly
The NIDDK conducts and supports clinical trials in many diseases and conditions, including endocrine diseases. The trials look to find new ways to prevent, detect, or treat disease and improve quality of life.
What are clinical trials for acromegaly?
Clinical trials—and other types of clinical studies—are part of medical research and involve people like you. When you volunteer to take part in a clinical study, you help doctors and researchers learn more about disease and improve health care for people in the future.
Researchers are studying many aspects of acromegaly and gigantism, such as
- use of medicine to treat gigantism in children and adolescents
- genetic factors that may cause pituitary tumors to develop, and how to treat the tumors and related complications in children and adults
Find out if clinical studies are right for you.
What clinical studies for acromegaly are looking for participants?
You can view a filtered list of clinical studies on acromegaly that are open and recruiting at
ClinicalTrials.gov. You can expand or narrow the list to include clinical studies from industry, universities, and individuals; however, the NIH does not review these studies and cannot ensure they are safe. Always talk with your health care provider before you participate in a clinical study.
References
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases
(NIDDK), part of the National Institutes of Health. NIDDK translates and disseminates research findings to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by NIDDK is carefully reviewed by NIDDK scientists and other experts.
The NIDDK would like to thank:
Lisa Nachtigall, M.D., Harvard Medical School and Massachusetts General Hospital Neuroendocrine and Pituitary Tumor Center