

Biliary Atresia
Return to Overview PageDefinition & Facts
In this section:
- What is biliary atresia?
- Are there different types of biliary atresia?
- How common is biliary atresia?
- Who is more likely to have biliary atresia?
- What are the complications of biliary atresia?
What is biliary atresia?
Biliary atresia is a condition in infants in which the bile ducts—tubes inside and outside the liver—are scarred and blocked. Bile ducts carry bile from the liver to the gallbladder for storage, and to the first part of the small intestine, also called the duodenum, for use in digestion. In infants with biliary atresia, bile can’t flow into the intestine, so bile builds up in the liver and damages it. The damage leads to scarring, loss of liver tissue and function, and cirrhosis.
Biliary atresia is life-threatening, but with treatment, most infants with biliary atresia survive to adulthood.
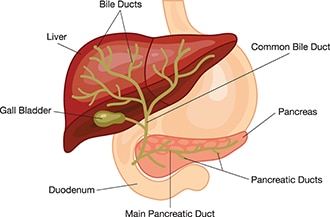
Are there different types of biliary atresia?
Doctors have identified different types of biliary atresia.
Biliary atresia without birth defects
In the most common type of biliary atresia, infants have no other major birth defects. Doctors may call this type of biliary atresia perinatal or isolated biliary atresia. A recent North American study found that 84 percent of infants with biliary atresia have this type.1
Biliary atresia with birth defects
Some infants have major birth defects—including problems with the heart, spleen, or intestines—along with biliary atresia. Doctors may call this fetal or embryonic biliary atresia. A recent North American study found that 16 percent of infants with biliary atresia have major birth defects.1
How common is biliary atresia?
Biliary atresia is rare and affects about 1 out of every 12,000 infants in the United States.2
Who is more likely to have biliary atresia?
Biliary atresia only occurs in newborn infants. The disease is slightly more common in female infants and in infants with Asian or African American heritage.3
What are the complications of biliary atresia?
Complications of biliary atresia include failure to thrive and malnutrition, cirrhosis and related complications, and liver failure.
Without treatment, infants with biliary atresia would develop cirrhosis within 6 months and liver failure within 1 year.3,4 By age 2, untreated infants would need a liver transplant to survive.4
Early treatment with a surgery called the Kasai procedure may slow or, in some cases, prevent the development of cirrhosis and liver failure. Even with treatment, about half of children with biliary atresia will need a liver transplant by age 2. Two-thirds will need a liver transplant sometime during childhood.3
Malnutrition
Even after treatment with the Kasai procedure, children with biliary atresia may have reduced bile flow to the small intestine and liver damage, leading to malnutrition and related problems with growth, such as failure to thrive. Read more about how biliary atresia affects nutrition.
Cirrhosis and related complications
Cirrhosis is a condition in which the liver breaks down and is unable to work normally. Scar tissue replaces healthy liver tissue, partly blocking the flow of blood through the liver. In the early stages of cirrhosis, the liver continues to work. As cirrhosis gets worse, the liver begins to fail.
In children with biliary atresia, cirrhosis may cause complications, including portal hypertension. Portal hypertension is high blood pressure in the portal vein, a blood vessel that carries blood from the intestines to the liver.
Portal hypertension may lead to specific complications, including
- a buildup of fluid in the abdomen, called ascites. Infection of this fluid can be very dangerous.
- enlarged blood vessels, called varices, which can develop in the esophagus, stomach, or both. Varices can break open and cause life-threatening bleeding in the digestive tract.
Liver failure
With liver failure, also called end-stage liver disease, the liver can no longer perform important functions or replace damaged cells. Infants and children with liver failure need a liver transplant to survive.
References
Symptoms & Causes
What are the symptoms of biliary atresia?
Typically, the first sign of biliary atresia is yellowing of the skin and whites of the eyes, called jaundice, which results from the buildup of bile in the body. Bile contains a reddish-yellow substance called bilirubin.
Infants often have jaundice in the first 2 weeks of life, so it is not easy to identify biliary atresia in newborn infants. Jaundice that lasts beyond 3 weeks of age may be the first sign of biliary atresia. Infants with biliary atresia typically develop jaundice by 3 to 6 weeks of age.
Infants with biliary atresia may also have pale yellow, gray, or white stools. Stools change color because bilirubin is not reaching the intestines and passing out of the body in the stool.

What causes biliary atresia?
Experts don’t know what causes biliary atresia. Research suggests that infants develop biliary atresia in the womb or shortly after birth. Experts are trying to find out if one or more of the following factors could play a role in causing biliary atresia:
- infections with certain viruses
- coming into contact with harmful chemicals
- problems with the immune system
- a problem during liver and bile duct development in the womb
- certain genes or changes in genes—called mutations—that may increase the chances of developing biliary atresia
Biliary atresia is not an inherited disease, meaning it does not pass from parent to child.
Diagnosis
How do doctors diagnose biliary atresia?
To diagnose biliary atresia, a doctor will ask about your infant’s medical and family history, perform a physical exam, and order a series of tests. Experts recommend testing for biliary atresia and other health problems in infants who still have jaundice 3 weeks after birth.
If test results suggest that an infant is likely to have biliary atresia, the next step is surgery to confirm the diagnosis.
Doctors may refer children with suspected biliary atresia to specialists, such as pediatric gastroenterologists, pediatric hepatologists, or pediatric surgeons.
Family and Medical history
The doctor will ask about your infant’s family and medical history. The doctor will also ask about symptoms such as jaundice and changes in stool color.
Physical exam
During a physical exam, the doctor may
- examine the infant’s body for signs of jaundice
- examine the infant’s body for other birth defects that sometimes occur along with biliary atresia
- feel the infant’s abdomen to check for an enlarged liver or spleen, which may be signs of biliary atresia
- check the color of the infant’s stool and urine

What tests do doctors use to diagnose biliary atresia?
Doctors may order some or all of the following tests to diagnose biliary atresia and rule out other health problems. Doctors may perform several tests because many other diseases can cause signs that are like the signs of biliary atresia.
Blood tests
A health care professional may take a blood sample from the infant and send the sample to a lab. Doctors may use blood tests to measure bilirubin levels and to check for signs of liver disease.
Ultrasound
Ultrasound uses a device called a transducer, which bounces safe, painless sound waves off organs to create images of their structure. Using ultrasound, doctors can rule out other health problems and look for signs that suggest an infant may have biliary atresia. However, an ultrasound cannot confirm a diagnosis of biliary atresia.
Hepatobiliary scan
A hepatobiliary scan is an imaging test that uses a small amount of safe radioactive material to create an image of the liver and bile ducts. The test can show if and where bile flow is blocked.
Liver biopsy
During a liver biopsy, a doctor will take pieces of tissue from the liver. A pathologist will examine the tissue under a microscope to look for signs of damage or disease. A liver biopsy can show whether an infant is likely to have biliary atresia. A biopsy can also help rule out or identify other liver problems.
How do doctors perform surgery to confirm the diagnosis of biliary atresia?
During diagnostic surgery, a pediatric surgeon makes a cut in the infant’s abdomen to directly examine the liver and bile ducts. Alternatively, surgeons may use a device called a laparoscope, which is inserted through a small incision and does not require the abdomen to be opened. If the surgeon confirms that the infant has biliary atresia, the surgeon will usually perform surgery to treat biliary atresia right away.
Treatment
How do doctors treat biliary atresia?
Doctors treat biliary atresia with a surgery called the Kasai procedure and eventually, in most cases, a liver transplant. Thanks to advances in treatment, more than 80 to 90 percent of infants with biliary atresia survive to adulthood.5,6
The Kasai procedure
The Kasai procedure is usually the first treatment for biliary atresia. The Kasai procedure does not cure biliary atresia. However, if the procedure is successful, it may slow liver damage and delay or prevent complications and the need for a liver transplant. The earlier the procedure is done, the more effective it may be.
During the procedure, a surgeon removes the damaged bile ducts outside the liver. The surgeon uses a loop of the infant’s own small intestine to replace the damaged bile ducts. If the surgery is successful, bile will flow directly from the liver to the small intestine. Within 3 months of the procedure, one has an idea of whether the surgery has worked or not. After a successful surgery, most infants no longer have jaundice and have a reduced risk of developing complications of advancing liver disease.
Complications. After the procedure, a common complication is infection of the liver, called cholangitis. Doctors may prescribe antibiotics after surgery to help prevent this infection. If cholangitis occurs, doctors treat it with antibiotics, usually intravenous (IV) antibiotics given in the hospital.
If the procedure is not successful, the flow of bile will remain blocked. After an unsuccessful procedure, infants will develop complications of biliary atresia and will usually need a liver transplant by age 2.5
Even after a successful surgery, most children will slowly develop complications of biliary atresia, over years or decades, and will eventually need a liver transplant. In some cases, after a successful procedure, children never need a liver transplant.
Liver transplant
If biliary atresia leads to serious complications, the infant or child will need a liver transplant. A liver transplant is surgery to remove a diseased or injured liver and replace it with a healthy liver from another person, called a donor.
Most children with biliary atresia eventually need a liver transplant, even after a successful Kasai procedure.
References
Eating, Diet, & Nutrition
How does biliary atresia affect nutrition?
Even after treatment with the Kasai procedure, children with biliary atresia may have reduced bile flow to the small intestine and liver damage, leading to
- problems digesting fats and absorbing fat-soluble vitamins
- loss of appetite
- a faster metabolism and a need for more calories
- low levels of protein, vitamins, and minerals
These problems may cause children with biliary atresia to become malnourished, and they may not grow normally.
What should infants and children with biliary atresia eat?
To make sure infants and children with biliary atresia get enough nutrients and calories, doctors may recommend
- a special eating plan
- a special formula for formula-fed infants
- supplements, which may be added to breast milk, formula, or food

Supplements for biliary atresia include vitamins—especially fat-soluble vitamins—and medium-chain triglyceride (MCT) oil. MCT oil adds calories to foods and is easier to digest without bile than other fats. Doctors may recommend other types of supplements as well.
If a child isn’t getting enough nutrients from food and supplements taken by mouth, a doctor may recommend using a feeding tube, called a nasogastric feeding tube, to provide high-calorie liquid directly to the stomach. In some cases, children with biliary atresia need to receive nutrition through an intravenous (IV) line. This type of feeding is called total parenteral nutrition (TPN).
Your child’s doctor or a dietitian can recommend a specific eating plan and supplements for your child.
What should infants and children eat after a liver transplant?
After a liver transplant, most infants and children can eat a healthy, balanced diet that is normal for their age. Learn more about living with a liver transplant.
Clinical Trials
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and other components of the National Institutes of Health (NIH) conduct and support research into many diseases and conditions.
What are clinical trials, and what role do children play in research?
Clinical trials are research studies involving people of all ages. Clinical trials look at new ways to prevent, detect, or treat disease. Researchers also use clinical trials to look at other aspects of care, such as improving quality of life. Research involving children helps scientists
- identify care that is best for a child
- find the best dose of medicines
- find treatments for conditions that only affect children
- treat conditions that behave differently in children
- understand how treatment affects a growing child’s body
Find out more about clinical trials and children.
Watch a video of NIDDK Director Dr. Griffin P. Rodgers explaining the importance of participating in clinical trials.
What clinical trials are open?
Clinical trials that are currently open and are recruiting can be viewed at ClinicalTrials.gov.
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases
(NIDDK), part of the National Institutes of Health. NIDDK translates and disseminates research findings to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by NIDDK is carefully reviewed by NIDDK scientists and other experts.
The NIDDK would like to thank:
Benjamin L. Shneider, M.D., Baylor College of Medicine, Texas Children’s Hospital