About Our Research
The purpose of our research is to understand how mutations of PKD genes cause polycystic kidney disease.
PKD is one of the most common, serious mendelian disorders of man and causes 4-5 percent of all end stage kidney disease in the United States. Affected individuals also suffer from other complications such as hypertension, cyst infections, abdominal pain, and have an increased risk of intracranial aneurysms. Present management is focused on treating symptoms. Understanding the mechanisms of disease will allow us to develop safe and effective treatments.
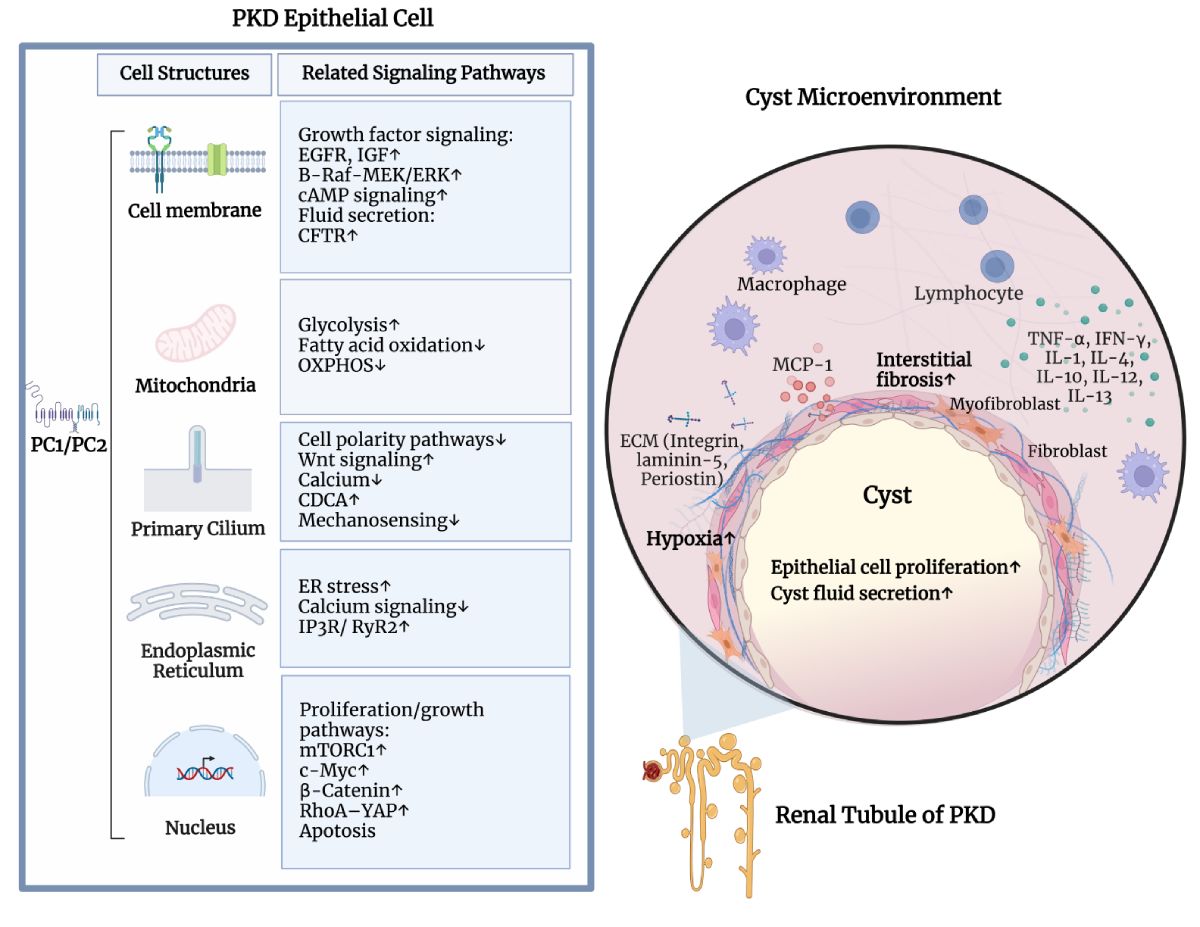
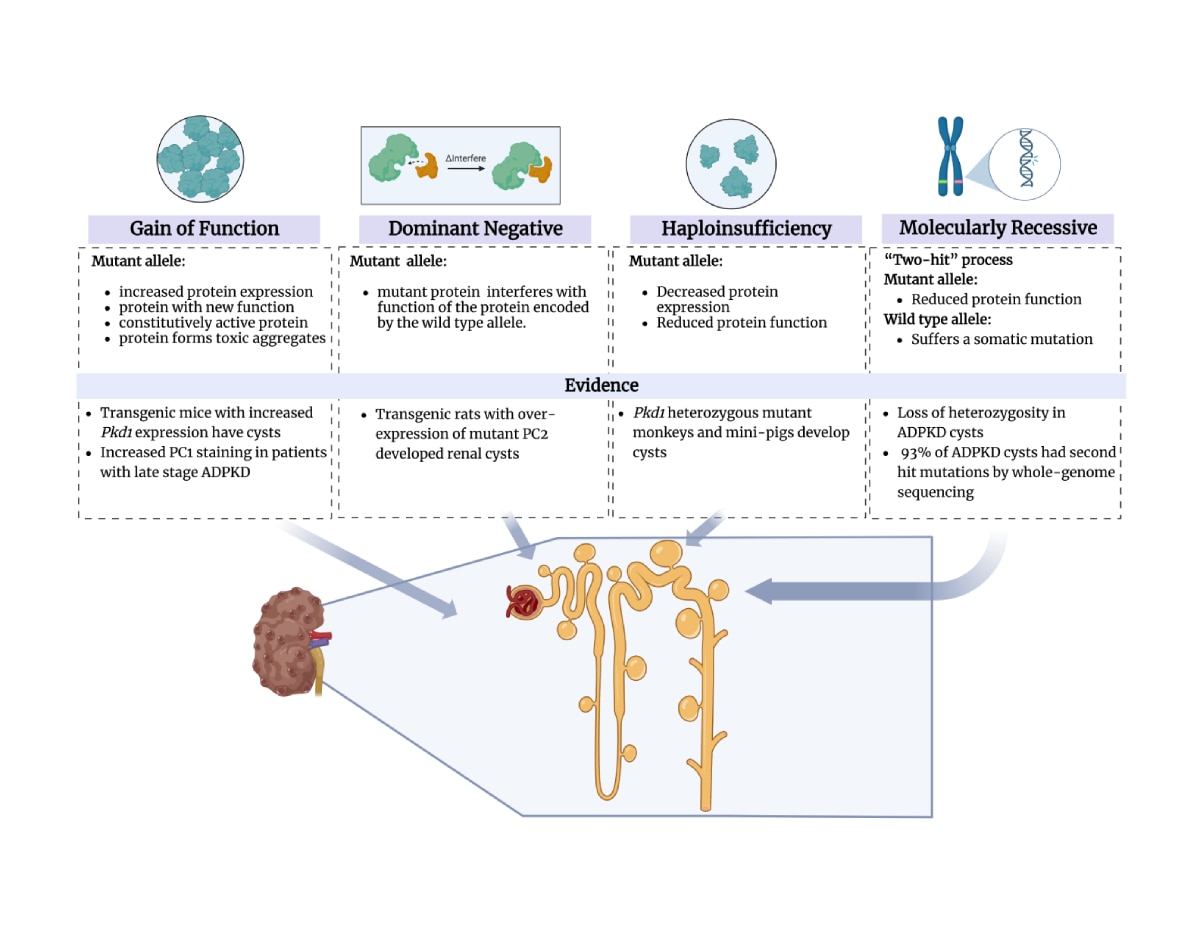
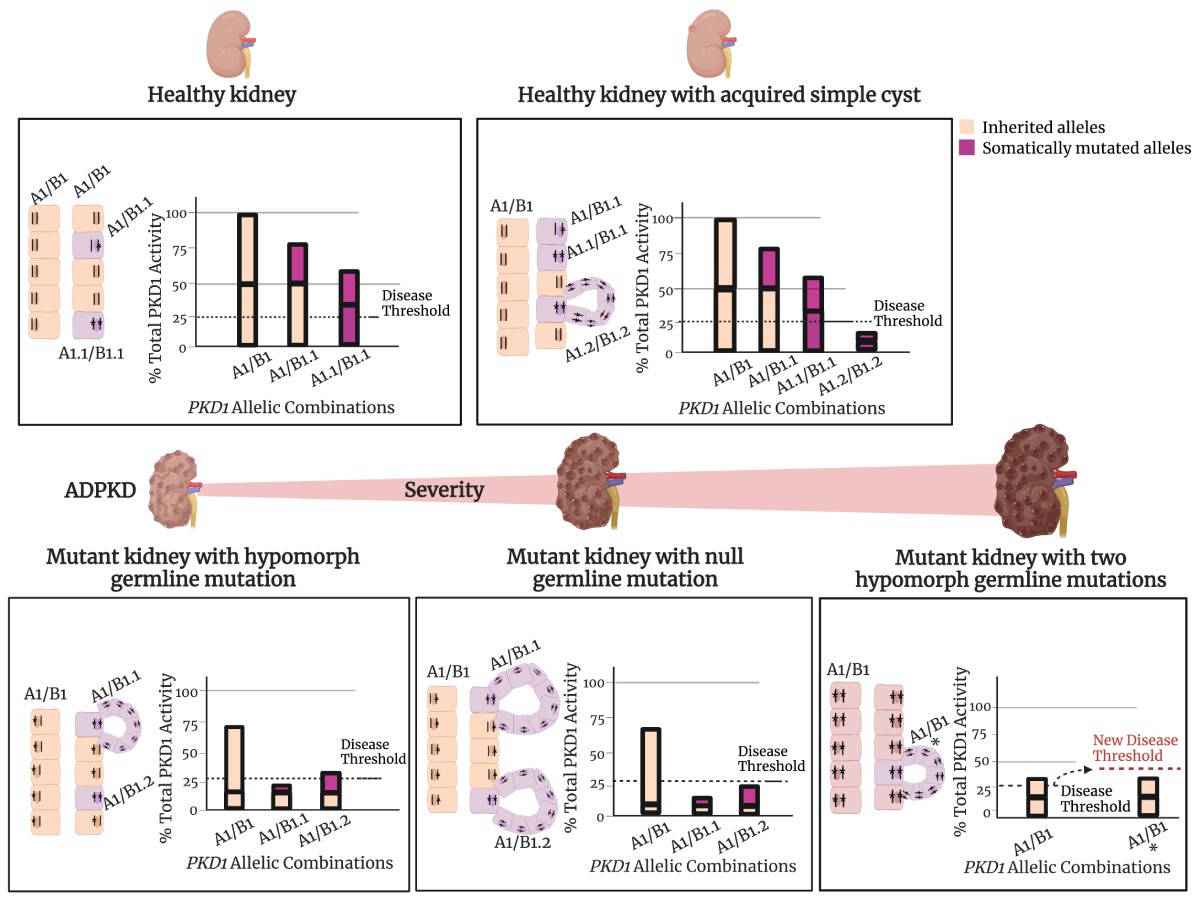
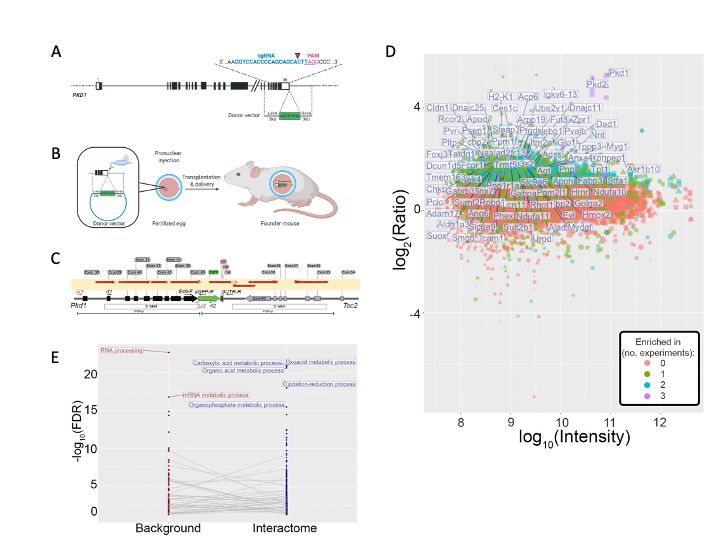
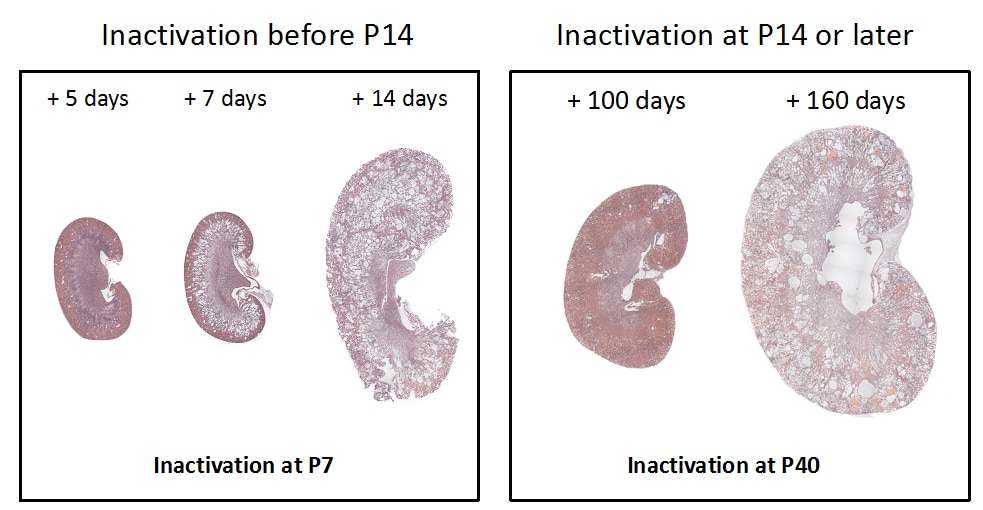
While the PKD gene products polycystin 1 (PC1) and polycystin 2 (PC2) are essential for establishing and maintaining normal tubule structure, we do not know how they do this. Several lines of evidence suggest that they function as a receptor-channel complex, but what they sense and signal is poorly understood. Though numerous pathways have been reported to be dysregulated in cystic epithelia, it is unclear how these link back to the function of the PC1/PC2 complex, which pathways are dysregulated at the earliest stages of cyst formation, and which are altered as secondary consequence of cystic dilations. We also do not understand why cysts form immediately after gene inactivation in young mice but take months to arise in older animals. What factors preserve tubular structure during this interval, and what triggers the subsequent failure? Understanding these processes is important not only from a pathobiological standpoint, but also to design more specific therapies. We also need better tools for assessing the progression of disease so we can better evaluate the effectiveness of clinical interventions.
Research Images