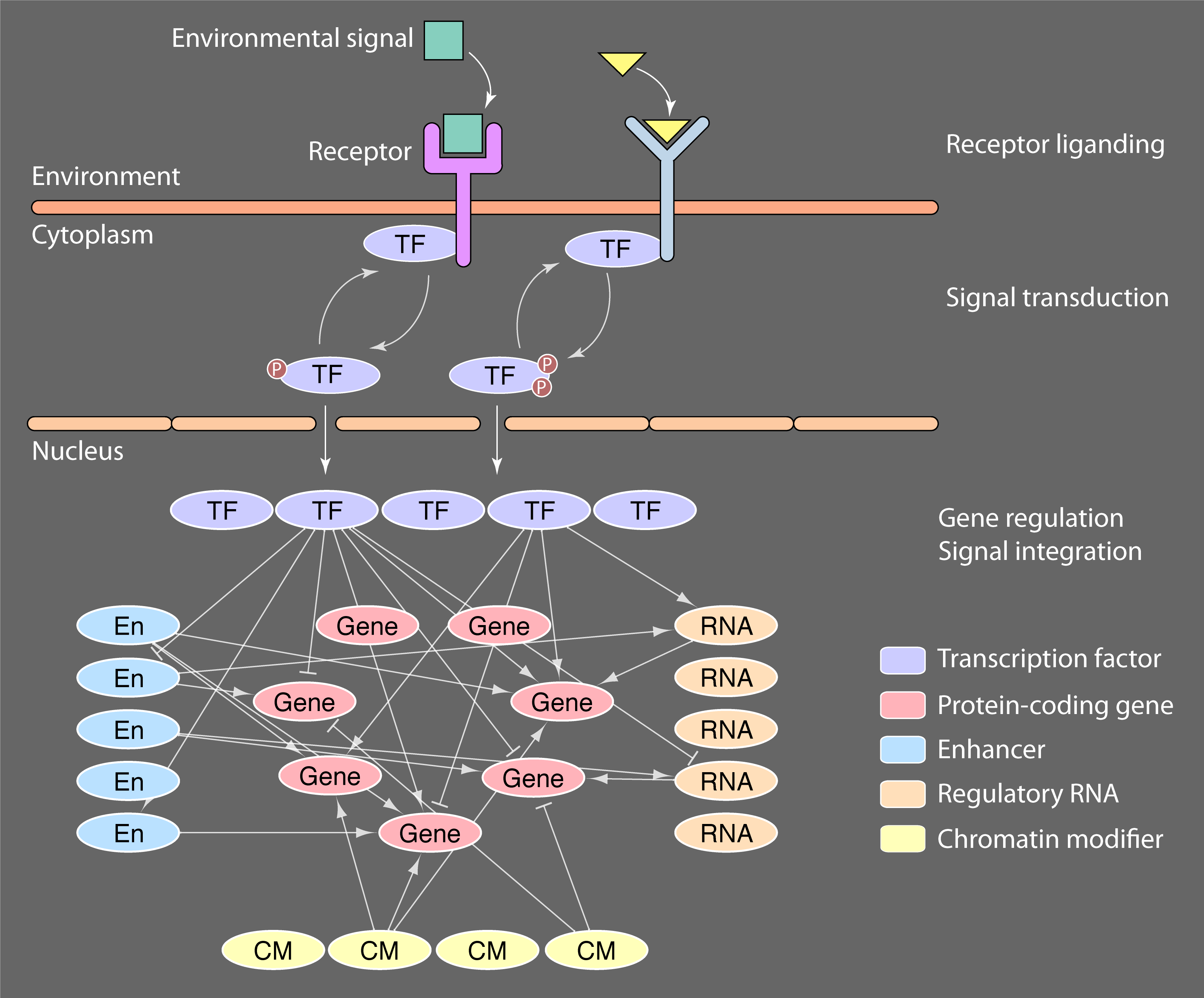
About Our Research
The immunoregulation section studies the basic mechanisms of tissue inflammation and its resolution. Our research focusses on the micro-environmental signals that drive inflammation in tissues and how these are integrated by networks of transcription factors in immune cells, such as T cells. By understanding the basic mechanisms of disease we endeavor to prevent tissue inflammation, accelerate its resolution and minimize loss of inflamed tissues from scarring.
Chronic inflammatory diseases, excluding infectious disease, account for more than 50% of deaths world-wide. The molecular mechanisms of tissue inflammation remain poorly understood. Moreover, there are no specific therapies to prevent tissue inflammation beyond broad-spectrum immunosuppressive drugs, which are non-specific and toxic, predisposing patients to infections and cancer development. When inflammation does resolve, there is often irreversible loss of function as a result of scarring processes initiated during the disease. The kidneys are exemplars of organs uniquely susceptible to autoimmune or inflammatory insults that result in progressive scarring and loss of function over time.
T cells are widely-recognized mediators of tissue inflammation. They are induced to differentiate from naïve precursors to either inflammatory or regulatory T cell lineages. The choice of differentiation pathway (“fate decisions”) are directed by environmental signals and interplay between a number of transcription factors. Once differentiated, T cells migrate to target tissues where they effect inflammation, regulation and tissue healing or healing through the expression of physical and soluble factors. Regulatory T cells (Tregs) are arguably the most important naturally-occurring anti-inflammatory cells in the body. They have highly potently immunosuppressive function and carry out a non-redundant role in preventing autoimmunity and in resolving inflammation. Mammals with loss of, or functional impairment in, these cells succumb to life-threatening multi-organ autoimmune diseases as a result of failure to regulate the immune system.
Research in the immunoregulation section is divided into two main themes:
- Understanding the origins and functions of microenvironmental signals that drive tissue inflammation; and
- Understanding how these are integrated by networks of transcription factors in T cells to determine inflammatory versus regulatory T cell differentiation and effector function.
In both themes our focus is to determine how transcription factors function within networks to initiate and drive gene regulation in both immune and non-immune cells within tissues such as the kidneys.
Applying Our Research
Our aim is to identify key nodes in the differentiation or function of T cells or kidney cells that can be therapeutically targeted by the development of novel treatments, in order to tip the balance between regulatory and inflammatory behavior and between tissue healing and scarring. We believe that successfully targeting these pathways could decrease inflammation or accelerate its resolution and thereby reduce the volume of kidney tissue lost due to scarring as inflammation is resolved.
Need for Further Study
These measures would reduce kidney injury, prolong the lifespan of injured kidneys, reduce the number of patients progressing to dialysis and increase the longevity of transplanted kidneys. By doing so the quality of life of patients that have had kidney injury will also be improved. All of these are areas in which new therapies with greater efficacy and reduced toxicity are currently required.
Research in Plain Language
Kidney disease is a common medical problem. We know that kidneys are often damaged by inflammation caused by the body’s own immune cells and that the treatments we have at present to reduce the inflammation are toxic. When inflammation does resolve, kidneys are very prone to developing scars that replace healthy tissue, so loss of normal kidney function progresses in the long-term, even if the original inflammation that started it has resolved. What we study is how the body’s immune cells take on inflammatory functions directed against the kidneys and how we can “re-program” them to take on anti-inflammatory properties instead. Simultaneously, we look at how the inflammation they cause results in kidney scarring. We hope to find ways in which we can disrupt this cycle and tip the balance towards anti-inflammation and reduced scarring.
Research Images