

Polycystic Kidney Disease (PKD)
View or Print All Sections
What is PKD?
Polycystic kidney disease is a genetic disorder that causes many cysts to grow in the kidneys. PKD cysts cause high blood pressure and problems with blood vessels in the brain and heart. Cysts in the liver can also occur with PKD.
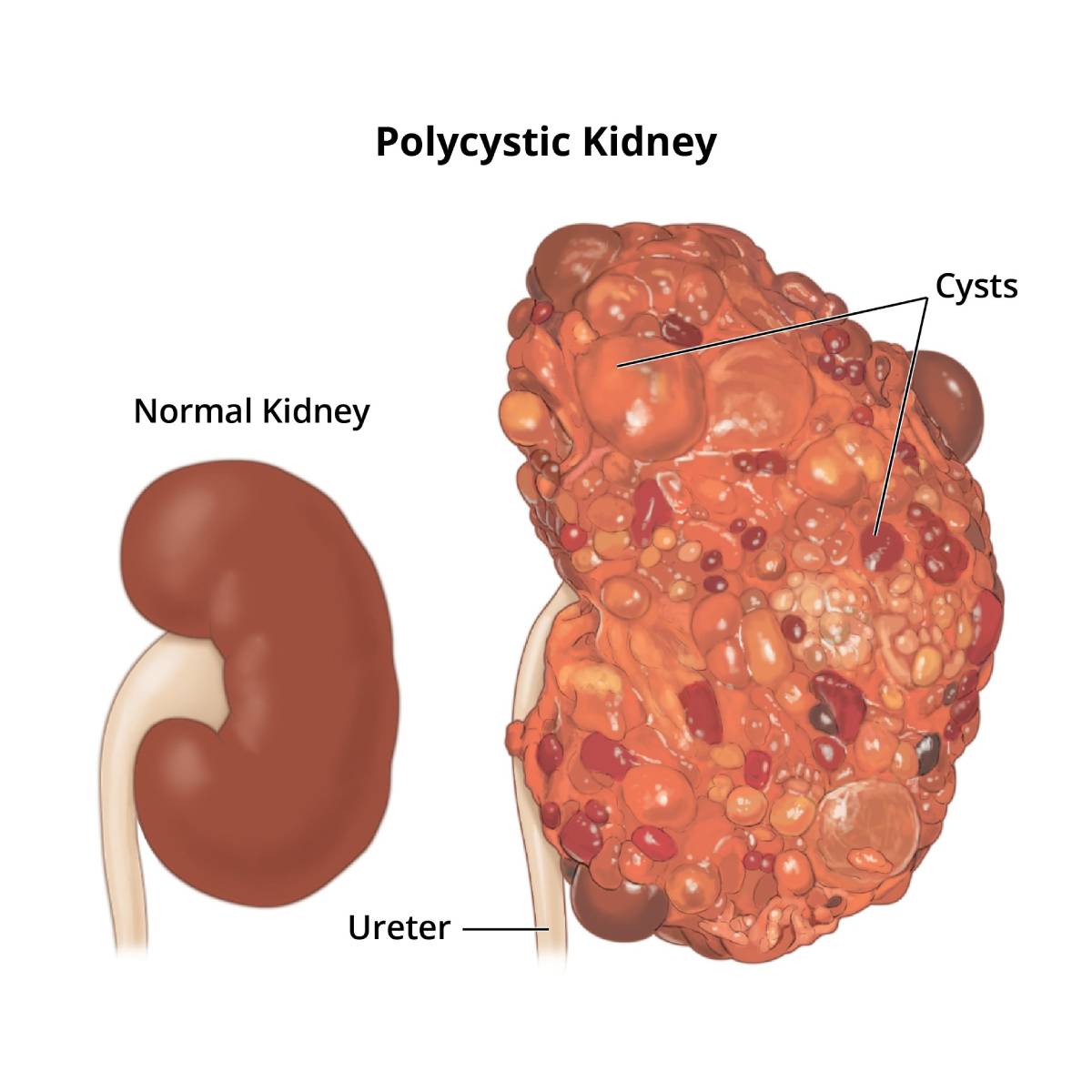
Autosomal Dominant PKD
Autosomal dominant PKD is often not diagnosed until later in adulthood. For this reason, health care providers often call autosomal dominant PKD "adult PKD." In many cases, PKD does not cause signs or symptoms until cysts are half an inch or larger.
Autosomal Recessive PKD
Autosomal recessive PKD is a rare genetic disorder that affects the liver as well as the kidneys. The signs of autosomal recessive PKD frequently appear in the earliest months of life, even in the womb, so health care providers often call it "infantile PKD."
Eating, Diet, & Nutrition for PKD
PKD may require changes in what you eat to control blood pressure. Following a healthy eating plan can help lower blood pressure. A health care provider may recommend the DASH eating plan, which focuses on fruits, vegetables, whole grains, and foods lower in sodium.
Related Conditions & Diseases
Kidney Failure
Clinical Trials
NIDDK and other components of the NIH conduct and support research into many diseases and conditions.
Related Research
See more about kidney diseases research at NIDDK.
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases
(NIDDK), part of the National Institutes of Health. NIDDK translates and disseminates research findings to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by NIDDK is carefully reviewed by NIDDK scientists and other experts.